![]()
Home Page: Taber home page.
![]()
![]()
Douglass F. Taber*
Yanong Wang
Department of Chemistry and Biochemistry
University of Delaware
Newark, DE 19176
Dedicated to John Alexander Oates, mentor and friend
Abstract
Diastereoselective and enantioselective hydrogenation of the racemic b-keto ester I to give the enantiomerically-pure (96% ee) ester II is reported. The conversion of the derived vinyl stannane III to the antibiotic marine alkaloid (-)-haliclonadiamine IV is described.
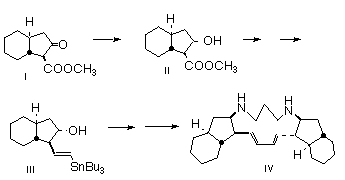
Introduction: (-)-Haliclonadiamine (1), a pentacyclic alkaloid isolated from the extract of a bright red encrusting marine sponge Haliclona sp. [1], collected near Palau, shows antifungal and antibiotic activity. Its structure and relative configuration were established by one and two-D NMR techniques and confirmed by X-ray analysis. The extract also contained papuamine (2 ), the epimer of 1, which had previously been isolated from an extract of Haliclona sp. [2] collected near Papua. We report the first stereocontrolled synthesis of (-)-haliclonadiamine (1).
At the inception of our work, no synthesis of papuamine or haliclonadiamine had been reported [3]. Our retrosynthetic analysis (Scheme 1) focused on vinyl stannane 3, which would have to be prepared in high enantiomeric and diastereomeric purity. We envisioned that 3 might be prepared from 4, which in turn could be available by Ru-BINAP hydrogenation of the racemic b-keto ester 5.
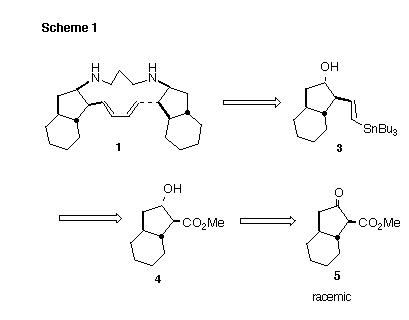
Synthesis: We had established that under equilibrating conditions, cyclozirconation (Scheme 2) of the inexpensive 1,7-octadiene proceeded to give cleanly the trans-fused diastereomer [4]. By combining this procedure with those developed for carbonylation of such zirconacycles [5], we were able to prepare the desired trans-fused ketone 7 in a single step. Carbomethoxylation of the symmetrical 7 then gave the racemic b-ketoester 5.
We were then faced with the problems of converting the racemic b-keto ester 5 into the enantiomerically pure and diastereomerically defined b-hydroxy ester 4. We thought that this might be accomplished by Ru-BINAP hydrogenation [6]. The reduction was known to give predominantly the trans b-hydroxy ester [7] when applied to a cyclic b-keto ester.
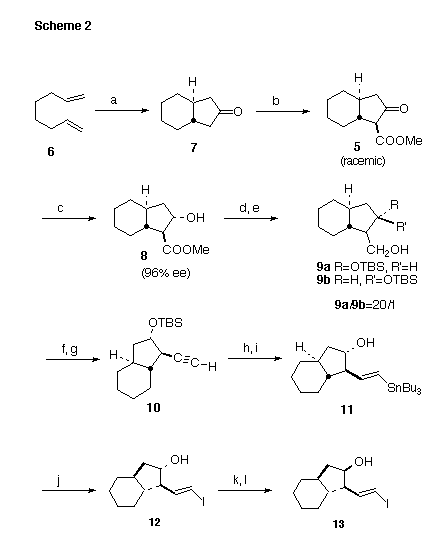
(a) Cp2ZrCl2/n-BuLi/PhCH3, 75 deg C, 3 hrs; CO, 24 hrs, -78 deg C-r.t.; HOAc, 79%. (b) (MeO)2CO/NaH/DME, 69%. (c) H2/Ru-BINAP/HCl/MeOH, 80-85 deg C, 87%. (d) TBDMSCl/imidazole/CH2Cl2, r.t.. (e) Dibal-H, 83% from 8. (f) (COCl)2/DMSO/CH2Cl2;. (g) (MeO)2P(O)C(=N2)COCH3/K2CO3/MeOH, 88% from 9a. (h) Dowex 50/MeOH. (j) n-Bu3SnH/AIBN, 87% from 10. (k) I2/Et2O, 96%. (l) 4-NO2C6H4CO2H/PPh3/DEAD/benzene. (m) K2CO3/MeOH, 79% from 12.
This reduction is thought to proceed by complexation of the intermediate Ru-H species with the ester carbonyl with subsequent delivery of the nucleophilic H to the ketone. As one enantiomer of the b-keto ester would fit the twist of the BINAP ligand and the other would not, we thought that one enantiomer of the ketone might reduce much more rapidly than the other.
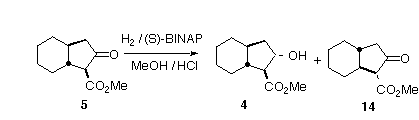
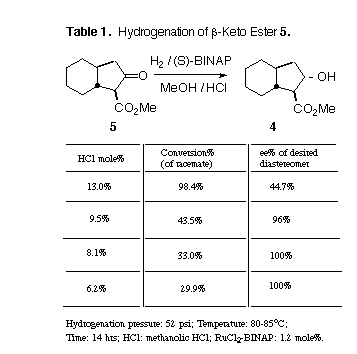
In the event (Table 1) we found that the racemic b-keto ester 5 only hydrogenated smoothly in the presence of added HCl [8]. By optimizing the amount of HCl added, we could control the proportion of total ketone reduced. We were pleased to observe that we could convert nearly 90% of the "matched" ketone without significant reduction of the other enantiomer. It is interesting that if too much HCl is added initially, the (S)-BINAP-RuCl2 will reduce the mismatched enantiomer also, to a mixture of diastereomers.
Silylation of the hydroxy ester 4 followed by reduction gave the primary alcohols 9a and 9b. The minor amount of cis alcohol produced in the reduction was removed at this stage. To establish the enantiomeric purity of 9a, we prepared the camphorsulfonate 15. The camphorsulfonate prepared from the racemic trans alcohol (from sodium borohydride reduction of the starting b-keto ester) showed nice resolution of the oxygenated carbons in the 13C NMR (74.0 d/73.8 d and 70.0 d/69.9 d). By this measurement, 9a was a 98:2 ratio of enantiomers.
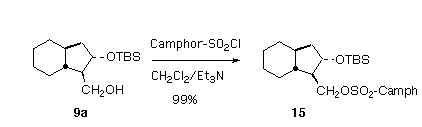
The alcohol 9a was oxidized to the aldehyde, which was homologated without purification by the Ohira procedure [9] to give the alkyne 10 . Deprotection of 10 followed by radical addition of tributyltin hydride [10] provided an 87% yield of the trans vinyl stannane 11. After converting stannane 11 to iodide 12 by exposure to I2 in ether[11] we inverted the hydroxy at C6. Thus, Mitsunobu coupling [12] of 12 with 4-nitrobenzoic acid followed by hydrolysis (K2CO3/methanol) provided the cis alcohol 13 in 75% overall yield.
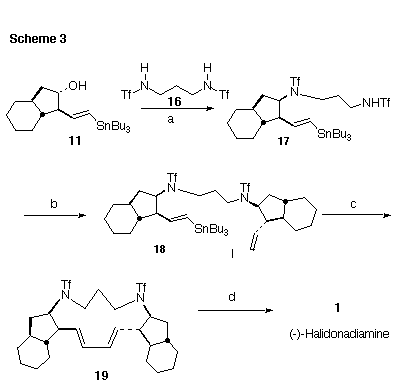
The next challenge we faced was construction of the N,N'-dialkyl 1,3-diamino-propane (Scheme 3). It seemed plausible that Mitsunobu coupling could be efficacious. The key to this approach was the selection of an activating group that would make the N-H sufficiently acidic to participate in the Mistsunobu reaction, while leaving the anion sufficiently nucleophilic. The activating group should also be easily removed. We found that the N, N'-bistriflamide [13] nicely met these criteria [14]. While the Mitsunobu coupling worked poorly in THF, reaction of 11 in benzene with two equivalents of N, N'-bistriflyl 1,3-diaminopropane (16) gave clean conversion to the monoalkylated diamide 17. The final Mitsunobu coupling of 17 with 13 proceeded smoothly in benzene to give the desired N,N'-dialkylated diamide 18.
Following the report by Barrett [3a] in the synthesis of (+)-papuamine, slow addition of 18 to 20 mol% of Pd(PPh3)4 at 100-105 deg C in toluene followed by 36 hours at reflux gave the cyclized product 19 in 43% yield. Deprotection of the bistriflamide 19 with LiAlH4 in ether [13] then gave the free base of (-)-haliclonadiamine (1). The 1H NMR, 13C NMR and MS of synthetic 1 were identical to the spectra of natural (-)-haliclonadiamine [1]. The 1H NMR was also identical with that recorded for a synthetic sample [3c].
(a) PPh3/DEAD/benzene, 66%. (b) 13/PPh3/DEAD/benzene, 56%. (c) (PPh3)4Pd/toluene, 100-105 deg C, 43%;. (d) LiAlH4/Et2O, sealed tube, 70 deg C, 34%.
Conclusion: During the course of this total synthesis of the unusual marine alkaloid (-)-haliclonadiamine (1), an efficient diastereoselective preparation of the b-keto ester 5 via intramolecular diene cyclozirconation and carbonylation was developed. The diastereoselective and enantioselective Ru-BINAP mediated hydrogenation of b-keto ester 5 to the b-hydroxy ester 4 was also achieved. Finally, the sequential Mitsunobu coupling with bistriflamide 16 as the nucleophile proved very efficient even with the sterically hindered secondary alcohols 16 and 13.
Acknowledgments: We thank the donors of the Petroleum Research Fund, administered by the American Chemical Society, for support of this work. We also thank Professor Clayton Heathcock for providing spectra of both natural and synthetic (-)-haliclonadiamine.
References and Notes:
(1) Fahy, E.; Molinski, T. F.; Harper, M. K.; Sullivan, B. W.; Faulkner, D. J.; Parkanyl, L.; Clardy, J. Tetrahedron Lett. 1988, 29, 3427.
(2) Baker, B. J.; Scheuer, P. J.; Shoolery, J. N. J. Am. Chem. Soc. 1988, 110, 965.
(3) Three enantioselective syntheses of papuamine 2 have been reported (a) Barrett, A. G. M.; Boys, M. L.; Boehm, T. L. J. Chem. Soc., Chem. Commun. 1994, 1881. (b) Weinreb, S. M.; Borzilleri, R. M.; Parvez, M. J. Am. Chem. Soc. 1994, 116, 9789; (c) McDermott, T. S.; Mortlock, A.; Heathcock, C. H. J. Org. Chem. 1996, 61, 700. (-)-Haliclonadiamine was prepared as a byproduct in this synthesis of (-)-papuamine.
(4) Taber. D. F.; Louey, J. P.; Wang, Y.; Nugent, W. A.; Dixon, D. A.; Harlow, R. L. J. Am. Chem. Soc. 1994, 116, 9457.
(5) (a) Rousset, C. J.; Swanson, D. R.; Lamaty, F.; Negichi, E-i. Tetrahedron Lett. 1989, 30, 5105. (b) Akita, M.; Yasuda, H.; Yamamoto, H.; Nakamura, A. Polyhedron 1991, 10 , 1.
(6) For the first report of the reduction of Ru-Binap mediated hydrogenation of b-keto esters to provide the b-hydroxy esters with excellent yield and enantioselectivity, see Noyori, R.; Ohkuma, T.; Kitamura, M. J. Am. Chem. Soc. 1987, 109, 5856.
(7) For further studies of Ru-Binap mediated hydrogenation of b-keto esters, see (a) Cesarotti, E.; Mauri, A.; Pallavicini, M.; Villa, L. Tetrahedron Lett. 1991, 32, 4381. (b) Kitamura, M.; Tokunaga, M.; Ohkuma, T.; Noyori, R. Tetrahedron Lett. 1991, 32, 4163. (c) Kitamura, M.; Tokunaga, M.; Noyori, R. J. Org. Chem. 1992, 57, 4053. (d) Taber, D. F.; Silverberg, L. J. Tetrahedron Lett. 1991, 32, 4227. (e) King, S. A.; Thompson, A. S.; King, A. O.; Verhoeven, T. R. J. Org. Chem. 1992, 57, 6689. (f) Kitamura, M.; Tokunaga, M.; Noyori, R. J. Am. Chem. Soc. 1993, 115, 144. (g) Genet, J. P.; Pinel, C.; Ratovelomanana-Vidal, V.; Mallart, S.; Pfister, X.; Cano De Andrade, M. C.; Laffitte, J. A. Tetrahedron 1994, 5, 665. (h) Mashima, K.; Kusano, K.; Sato, N.; Matsumura, Y.; Nozaki, K.; Kumobayashi, H.; Sayo, N.; Hori, Y.; Ishizaki, T.; Akutagawa, S.; Takaya, H. J. Org. Chem. 1994, 59, 3064. (i) Burk, M. J.; Harper, T. G. P.; Kalberg, C. S. J. Am. Chem. Soc. 1995, 117, 4423.
(8) For the role of added acid in Ru-BINAP hydrogenation, see ref. (7d) and (7e).
(9) Ohira, S. Syn. Comm. 1989, 19, 561.
(10) (a) Stille. J. K. Angew. Chem. Int. Ed. Engl. 1986, 25, 508. (b) Zhang, H. X.; Guibe, F.; Balavoine, G. J. Org. Chem. 1990, 55, 1857.
(11) Hanson, R. N.; El-Wakil, H. J. Org. Chem. 1987, 52, 3687.
(12) (a) Maruyama, H.; Hiraoka, T. J. Org. Chem. 1986, 51, 399. (b) Martin, S. F.; Dodge, J. A. Tetrahedron Lett. 1991, 32, 3017. (c) Caine, D.; Kotian, P. L. J. Org. Chem. 1992, 57, 6587.
(13) (a) Hendrickson, J. B.; Bergeron, R. Tetrahedron Lett. 1973, 3839. (b) Hendrickson, J. B.; Bergeron, R.; Sternbach, D. J. Am. Chem. Soc. 1973, 95, 3412.
(14) After this work was completed, an independent report of efficient Mitsunobu coupling of an alkyl triflamide appeared: Bell, K.E.; Knight, D.W.; Gravestock, M.B. Tetrahedron Lett. 1995, 36, 8681.
Questions and comments to Douglass F. Taber