![]()
Douglass F. Taber . . . U. of Delaware
Home Page: Taber home page.
![]()
Highly Diastereoselective Cyclopentane Construction: Enantioselective Synthesis of the Dendrobatid Alkaloid 251F
Douglass F. Taber*
Kamfia K. You
Department of Chemistry and Biochemistry
University of Delaware
Newark, DE 19716
Abstract
Rhodium-mediated cyclization of diazo ester I proceeds with high diastereoselectivity, to give the tetrasubstituted cyclopentane II. This cyclization is a key step in the enantioselective synthesis of the dendrobatid alkaloid 251F III.
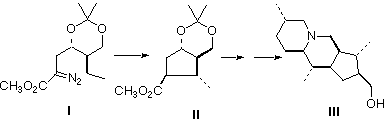
Introduction: In 1992 Daly and Spande reported [1] the isolation and structural elucidation, primarily by high field 1H and 13C NMR and mass spectrometry, of alkaloid 251F (1) from the skin exudate of the dendrobatid poison frog Minyobates bombetes of Colombia. Unlike most of the dendrobatid alkaloids, which are apparently acetogenins, 1 is clearly terpene-derived. We report the first total synthesis, and thus structural confirmation, of 1, based on the retrosynthetic analysis illustrated. The most critical observations in this synthesis are that both the Rh-mediated cyclization of 4 and the anionic cyclization of 2 can be effected with excellent diastereoselectivity.
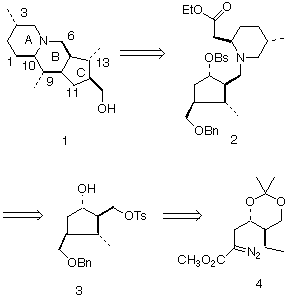
Preparation of the C Ring: For the proposed convergent assembly to succeed, it was necessary to prepare both the carbocyclic C ring and the piperidine A ring in high enantiomeric purity. While a variety of approaches to enantiomerically pure cyclopentanes have been developed [2 ], none of these is readily applicable to the preparation of such a highly substituted cyclopentane as 3. We therefore investigated the Rh-mediated cyclization [3,4] of diazoester 4(Scheme 1).
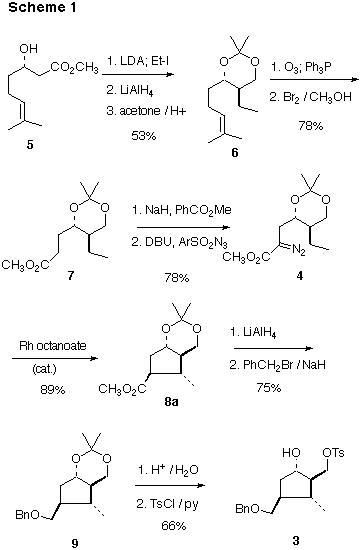
We were attracted to this approach by the our already-established preparation [5] of the enantiomerically pure -hydroxy ester 5 (two steps from the inexpensive 6-methyl-5-heptene-2-one, 90% overall yield and 97% e.e., by Ru BINAP hydrogenation [6] of the intermediate -ketoester). Alkylation [7] of the dianion proceeded to give the expected anti product, which was reduced and protected to afford 6. Ozonolysis of the alkene [6] followed by oxidation [8] gave the ester 7.
Evans reported traces of diazo transfer on reaction of 4-nitrobenzenesulfonylazide with an ester enolate [9]. We have found that initial benzoylation of the ester enolate substantially increases the yield of the subsequent diazo transfer. This is the first practical procedure for direct diazo transfer to an ester [10].
Cyclization of 4 could proceed to a give a mixture of one or more of the diastereomers 8a - 8d (Scheme 2). There are four corresponding diastereomeric transition states, 4a - 4d, leading to cyclization. Previous work on Rh-mediated intramolecular C-H insertion [3,4] supports the concept that initial complexation of the intermediate Rh carbene with the target C-H bond is rapid and reversible. We reasoned that bridging the 1,3-diol with the acetonide protecting group could provide a rigidity to these transition states.
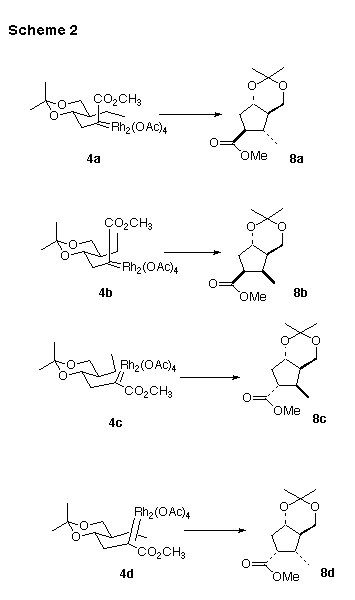
Considering each of the diastereomeric transition states in turn, 4a seemed the most favorable. Transition state 4b looks very much like 4a, with, however, an additional destabilizing buttressing interaction between the methyl group and the ester. In transition state 4c, the Rh dimer, swung out of the way in 4a and 4b, is tucked up in a sterically more congested area under the ring. Transition state 4d has the same problem, and also adds the buttressing between the methyl group and the ester seen in 4b [11].
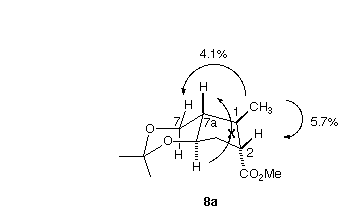
In fact, the cyclization of 4proceeded smoothly, to give 8a as a single dominant (13C NMR) diastereomer. The relative configuration of 8a was assigned by a combination of COESY and NOE techniques. The most significant observations were a 4.1% NOE between the methyl group and the equatorial H at C-7, and a 5.7% NOE between the methyl group and the H at C-2. The lack of an NOE between the ring fusion H's confirmed the trans ring fusion. Given the rigid nature of the chair conformation of the six-membered ring, these observations then secure the relative configuration of 8a.
Reduction of the ester and subsequent protection led to 9. Monotosylation of the derived diol then gave 3, in twelve steps and 14% overall yield from 5. Using this approach, we have routinely prepared gram quantities of enantiomerically pure 3.
Preparation of the Piperidine A Ring: To pursue the proposed convergent assembly of B>1, we also needed gram quantities of the enantiomerically pure piperidine B>14 (Scheme 3). This was conveniently available starting with the Sharpless asymmetric epoxidation [12] of geraniol 10. Reduction of the epoxide (92% e.e.) following the Hutchins procedure [13] proceeded cleanly to give the 2-hydroxycitronellol 11 [14], which on periodate cleavage [15] followed by reductive workup gave norcitronellol 12. We have found this assembly of 12 to be much more convenient than alternative chiral auxiliary-based methods.
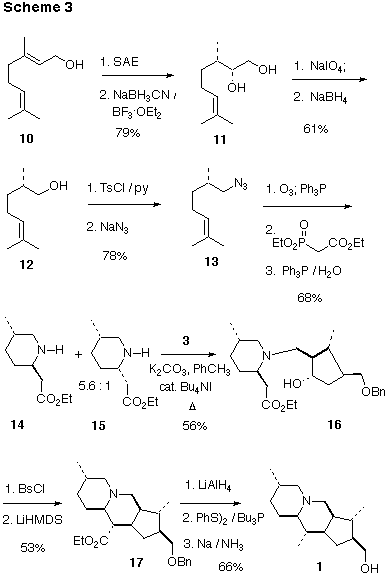
Ozonolysis of the unstable azide 13 followed by phosphonate condensation and reduction of the azide [16] at -50 afforded 14 and 15 in a ratio of 5.6 : 1. Assignment of the relative configuration of 14 and 15 was made by comparison of 1H NMR chemical shifts with those for known substituted piperidines [17]. At 0 degrees, the same cyclization proceeded to give 14 and 15 in a ratio of 1.1 : 1.
Convergent Assembly of 1: Alkylation of 14 with 3 proceeded smoothly, to give 16 . There were then two uncertainties to be faced in approaching the proposed intramolecular alkylation to close the B ring. First, it would be necessary to purify the unstable amino benzenesulfonate derived from 16 . Even if the benzenesulfonate could be sufficiently purified, the attempted enolate formation might result instead in -elimination.
Direct formation of the cis-fused azetidinium salt was indeed a real hazard. We observed that the isolated yield of the benzenesulfonate dropped off quickly with extended reaction time. Nevertheless, rapid preparation and purification allowed the isolation of the desired benzenesulfonate.
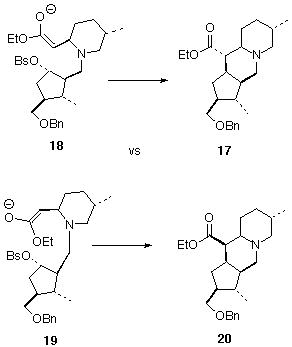
The demonstration by Still [18] that it is possible to generate and alkylate the enolate of a b-amino ester made cyclization plausible. The question of the relative configuration of the newly-established stereogenic center remained. We reasoned that transition state 18 would be less congested than 19 , so 17 would be favored over 20. While cyclization proceeded smoothly, to give 17 as a single dominant diastereomer, it is not impossible that any of ester 20 that formed could have been equilibrated to 17 under the conditions of the cyclization.
To complete the synthesis, it was necessary to convert the ester to a methyl group. Several methods [19] have been put forward for effecting this transformation. We have developed what promises to be an efficient alternative. Thus, ester 17 was reduced to the corresponding alcohol, which was then converted to the sulfide [20]. Dissolving metal reduction [21] then effected clean desulfurization as well as debenzylation, to give 1.
The amino alcohol from the reduction had a mass spectrum congruent with that reported for 1. The identity of the synthetic amino alcohol with the natural alkaloid was confirmed by 1H and 13C NMR [22], GC-MS coinjection on a capillary GC column, and GC-IR [23].
Conclusion: The isolation and structure of 1 was carried out with 300 g of material, the total that had been purified from the Minyobates bombetes extract [1]. The convergent assembly of 1 outlined here, even in its initial form, has already increased the supply of the purified alkaloid by a factor of more than one hundred. The high diastereoselectivity observed for the cyclization of 4is especially noteworthy. Our preliminary investigations with additional a -diazo esters indicate that these substrates often cyclize with high diastereoselectivity [11].
Acknowledgments: We thank M.P. Doyle and T.F. Spande for helpful discussions, and Zeneca Pharmaceuticals for financial support.
References
(1) Spande, T.F.; Garraffo, H.M.; Yeh, H.J.C.; Pu, Q.L.; Pannell, L.K.; Daly, J.W. J. Nat. Prod. 1992, 55, 822.
(2) For alternative methods for the enantioselective construction of highly substituted cyclopentanes, see (a) Allan, R.D.; Johnston, G.A.R.; Twitchin, B. Aust. J. Chem. 1979, 32, 2517. (b) Colombo, L.; Gennari, C.; Resnati, G.; Scolastico, C. Synthesis, 1981, 74. (c) Klunder, A.J.H.; Huizinga, W.B.; Sessnik, P.J.M.; Zwanenburg, B. Tetrahedron Lett. 1987, 28, 357. (d) Trigalo, F.; Buisson, D.; Azerad, R. Tetrahedron Lett. 1988, 29, 6109. (e) Henly, R.; Elie, C.J.J.; Buser, H.P.; Ramos, G.; Moser, H.E. Tetrahedron Lett. 1993, 34, 2923.
(3) For the first observation of the efficient cyclization of simple -diazo esters, see Taber, D.F.; Hennessy, M.J.; Louey, J.P. J. Org. Chem. 1992, 57, 436.
(4) For leading references to Rh-mediated intramolecular C-H insertion, see (a) Doyle, M.P.; Dyatkin, A.B.; Roos, G.H.P.; Canas, F.; Pierson, D.A.; van Basten, A.; Mller, P.; Polleux, P. J. Am. Chem. Soc. 1994, 116, 4507. (b) Wang, P.; Adams, J. J. Am. Chem. Soc. 1994, 116, 3296. (c) Doyle, M.P. In Homogeneous Transition Metal Catalysts in Organic Synthesis; Moser, W.R., Slocum, D.W., Eds.; ACS Advanced Chemistry Series 230; American Chemical Society, Washington, D.C., 1992, Chp. 30. (d) Taber, D.F. Comprehensive Organic Synthesis, V. 3, Pattenden, G. Ed., Pergamon Press, Oxford, 1991, p. 1045.
(5) Taber, D.F.; Silverberg, L.J.; Robinson, E.D. J. Am. Chem. Soc. 1991, 113, 6639.
(6) (a) Noyori, R.; Ohkuma, T.; Kitamura, M.; Takaya, H.; Sayo, N.; Kumobayashi, H.; Akutagawa, A. J. Am. Chem.Soc. 1987, 109, 5856. (b) Kitamura, M.; Ohkuma, T.; Inoue, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, A.; Ohta, T.; Takaya, H.; Noyori, R. J. Am. Chem.Soc. 1988, 110, 629. (c) Kitamura, M.; Ohkuma, T.; Takaya, H.; Noyori, R. Tetrahedron Lett. 1988, 29, 1555. (d) Nishi, T.; Kitamura, M.; Ohkuma, T.; Noyori, R. Tetrahedron Lett. 1988, 29, 6327. (e) Schreiber, S.L.; Kelly, S.E.; Porco, J.A.; Sammakia, T.; Suh, E.M. J. Am. Chem. Soc. 1988, 110, 6210. (f) Noyori, R.; Ikeda, T.; Ohkuma, T.; Widhalm, M.; Kitamura, M.; Takaya, H.; Akutagawa, S.; Sayo, N.; Saito, T.; Taketomi, T.; Kumobayashi, H. J. Am. Chem. Soc. 1989, 111, 9134. (g) Jones, B.A.; Yamaguchi, M.; Patten, A.; Danishefsky, S.J.; Ragan, J.A.; Smith, D.B.; Schreiber, S.L. J. Org. Chem. 1989, 54, 17. (h) Oppolzer, W.; Wills, M.; Starkmann, C.; Bernardinelli, G. Tetrahedron Lett. 1990, 31, 4117.
(7) A 95:5 ratio is reported for the alkylation of an analogous -alkoxy ester enolate: Frater, G. Helv. Chim. Acta 1979, 62, 2825. We have not yet been able to isolate or characterize the very minor diastereomer of 7. The yield reported for 7 is for diastereomerically pure (13C) material.
(8) Williams, D.R.; Klingler, F.D.; Allen, E.E.; Lichtenthaler, F.W. Tetrahedron Lett. 1988, 29, 5087.
(9) Evans, D.A.; Britton, T.C.; Ellman, J.A.; Dorow, R.L. J. Am. Chem. Soc. 1990, 112, 4011.
(10) For an overview of diazo transfer chemistry, see (a) Regitz, M.; Maas, G. Diazo Compounds: Properties and Synthesis, Academic Press, Orlando, 1986. (b) Askani, R; Taber, D.F. Comprehensive Organic Synthesis, Vol. 6, E. Winterfeldt, Ed., Pergamon, Oxford, 1991, p. 103.
(11) We have developed a computational approach that rationalizes both the highly diastereoselective cyclization of 4, and the similarly diastereoselective cyclization of other substituted a-diazo esters: Taber, D.F.; You, K.K.; Rheingold, A.L. J. Am. Chem. Soc. 1996, 118, 547.
(12) (a) Katsuki, T.; Sharpless, K.B. J. Am. Chem. Soc. 1980, 102, 5974. (b) Hanson, R.M.; Sharpless, K.B. J. Org. Chem. 1986, 51, 1922.
(13) Hutchins, R.O.; Taffer, I.M.; Burgoyne, W. J. Org. Chem. 1981, 46, 5214.
(14) Taber, D.F.; Houze, J. B. J. Org. Chem. 1994, 59, 4004.
(15) Daumas, M.; Vo-Quang,Y.; Vo-Quang,L.; Le Goffic, F. Synthesis 1989, 64.
(16) Knouzi, N.; Vaultier, M.; Toupet, L.; Carrie, R. Tetrahedron Lett. 1987, 28, 1757.
(17) Cahill, R.; Crabb, T.A. Org. Mag. Res. 1972, 4, 259.
(18) For an earlier use of a -amino ester enolate in synthesis, see Still, W.C.; Schneider, M.J. J. Am. Chem. Soc. 1977, 99, 948.
(19) For a review of methods for the deoxygenation of alcohols, see (a) Hartwig, W. Tetrahedron 1983, 39, 2609. For additional procedures for the deoxygenation of a primary alcohol to a methyl group, see (b) Barton, D.H.R.; Motherwell, W.B,.; Stange, A. Synthesis 1981, 743. (c) Trost, B.M.; Renaut, P. J. Am. Chem. Soc. 1982, 104, 6668. (d) Grether, G.;Mitt, T.; Williams, T.H.; Uskokovic, M.R. J. Org. Chem. 1983, 48, 5309. (e) Feldman, K.S.; Wu, M.-J.; Rotella, D.P. J. Am. Chem. Soc. 1990, 112, 8490. (f) Barton. D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Tetrahedron Lett. 1990, 31, 4681. (g) Barton. D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Tetrahedron Lett. 1992, 33, 2311. (h) Barton. D.H.R.; Jang, D.O.; Jaszberenyi, J.C. Tetrahedron Lett. 1990, 31, 4681.
(20) Cleary, D.G. Syn. Comm. 1989, 19, 737.
(21) For the dissolving metal reduction of secondary phenyl alkyl thioethers to alkanes, see (a) Haskell, T.H.; Woo, P.W.K.; Watson, D.R. J. Org. Chem. 1977, 42, 1302. (b) Masaki, Y.; Hashimoto, K.; Sakuma, K.; Kaji, K. Tetrahedron Lett. 1982, 23, 1481. (c) Suridov, A.F.; Ermolenko, M.S.; Yashunsky, D.V.; Borodkin, V.S.; Kochetkov, N.D. Tetrahedron Lett. 1987, 28, 3835.
(22) The 1H and 13C spectra were acquired in D2O/DCl.
(23) We thank T.F. Spande, H.M. Garraffo and H.C.J. Yeh, Laboratory of Bioorganic Chemistry, NIH, for making these comparisons.
(24) Hashimoto, S.-1.; Watanabe, N.; Sato, T.; Shiro, M.; Ikegami, S. Tetrahedron Lett. 1993, 33, 5109.
![]()
Questions and comments to Douglass F.
Taber