
Douglass F. Taber . . . U. of Delaware
Home Page: Taber home page.

Synthesis of Mesembrine
Intramolecular alkylidene C-H insertion is a key step in our recent synthesis of mesembrine.
Synthesis of (+)-Cassiol
Douglass F. Taber*
Robert P. Meagley
Douglas J. Doren
Department of Chemistry and Biochemistry
University of Delaware
Newark, DE 19716
Abstract
C-H insertion of the alkylidene carbene II generated from ketone I proceeds stereospecifically to give the cyclopentene. This cyclization is a key step in the enantioselective synthesis of (+)-cassiol IV . Two methods for converting the ketone to the alkylidene carbene are presented.
.
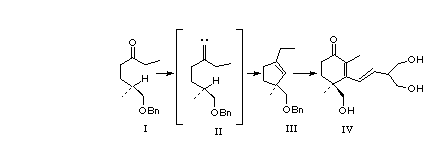
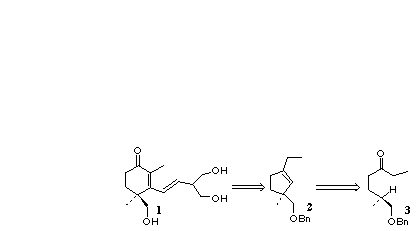
Introduction: The stereospecific construction of functionalized five and six membered carbocycles continues to challenge practitioners of organic synthesis. The work of Gilbert [1a, b] and later of Ohira [1c, d] suggested that the intramolecular C-H insertion of an alkylidene carbene1 could offer a general method for converting acyclic ternary stereogenic centers into cyclic quaternary centers of defined absolute configuration. We detail here our exploration of several procedures for generating alkylidene carbenes from ketones, and our delineation of experimentally effective protocols for effecting these cyclizations. The utility of this approach is illustrated by an expeditious synthesis of (+)-cassiol (1), a potent anti-ulcerant isolated from Cinnimomum cassia, from the enantiomerically pure ketone 3 via the cyclopentene 2[ 2].
Background: The transition state for C-H insertion by alkylidene carbene
From a theoretical standpoint, the alkylidene carbene is an interesting species, bearing both similarities to alkyl carbenoids, and significant differences. Ab initio calculations3a employing a 6-31G** basis set on 4 indicate that this is a singlet carbene,
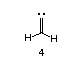
with hybridization midway between sp2 and sp. Experimental evidence for a singlet state may be found in earlier work by Gilbert and Ohira who have confirmed that C-H insertion into a stereochemically defined methine proceeded with retention of absolute configuration [1].
A key issue that had not been resolved was one of diastereoselectivity. Alkyl carbenoids are known to effect C-H insertion to form cyclopentanes with significant diastereoselectivity [4 ]. We postulated that though more reactive, the alkylidene carbene could show a similar diastereomeric preference. The transition state geometry for the C-H insertion may be represented as a half chair.
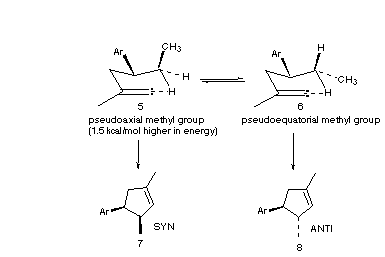
In this example, the phenyl substituent lies in a pseudoequatorial position. The methyl group adjacent to the phenyl group may either be pseudoequatorial or pseudoaxial. In each case only one of the two possible methylene C-H bond molecular orbitals is periplanar with and therefore can overlap with the empty orbital of the alkylidene carbene. The case where the methyl group is pseudoaxial leads to the syn diastereomer, and the case where the methyl group is pseudoequatorial leads to the anti diastereomer. A Molecular Mechanics [3b] simulation of this system with an MM2 force field and a "weak bond" between the alkylidene carbon and the target H indicates that at the point of commitment, the syn transition state lies 1.5 kcal/mol higher in energy than the anti transition state, largely due to 1, 3 diaxial interactions with the methyl group. Based on this model, we predicted that the anti product would be the major diastereomer formed in the reaction.
Alkylidene carbene generation by way of the cumulated diazo alkene:
To test the above hypothesis, we prepared ketone 9 from 4-(4-(N,N-dimethylamino)phenyl)-3-butene-2-one by copper mediated conjugate addition of ethylmagnesium bromide. We expected that this would be a challenging substrate to cyclize, as the inherently slower rate for methylene compared to methine insertion would be exacerbated by inductive electron withdrawal by the adjacent aromatic ring. Thus, alternative pathways could intervene [1a].
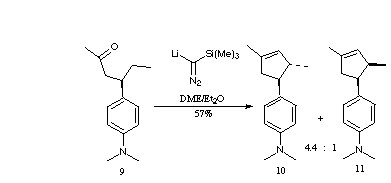
Initially, we explored procedures that would convert ketones into alkylidene carbenes directly. The use of the anion of dimethyl (diazomethylphosphonate) to effect this transformation was pioneered by Gilbert [1a,b]. This reagent is not commercially available, but can be prepared from dimethyl(phthalimidomethylphosphonate) in two steps. We have found Ohira's substitution of trimethylsilyldiazomethane in this reaction to be more convenient, since this reagent is commercially available [1c-f].
We initially observed that the Ohira procedure, as published, was not effective for the C-H insertion into deactivated methine and methylene bonds. Selection of the solvent system proved to be critical to success [6]. It was crucial to employ DME for smooth insertion into the inherently less reactive methylene C-H bonds. Further, it was also determined that the system would not tolerate hexane, either as a solvent for the commercial trimethylsilyldiazomethane, or as a solvent for the base (i.e. n-butyl lithium). We therefore employed methyl lithium, available as a salt free solution in diethyl ether, and carefully distilled the trimethylsilyldiazomethane away from the hexane solvent [7]. Treatment of the DME solution of trimethylsilyldiazomethane with methyl lithium at -78 resulted in a slurry of trimethylsilyldiazomethyl lithium that efficiently converted 9 to a 4.4:1 mixture of 10 and 11.
Intramolecular Alkylidene Insertion: Methine Insertion
In support of our continuing studies toward the synthesis of taxol, we next investigated the preparation of enone 14, a possible taxol A-ring synthon [1f]. Crucial to this effort is the conversion of ketone 12 to cyclopentene 13 via the kinetically favorable alkylidene carbene methine insertion reaction. Having accomplished the C-H insertion into the electronically deactivated methylene of 9, we expected that application of our methodology to this system would pose little difficulty, for in addition to being a methine, the target C-H bond is further activated for carbene insertion by a-oxygenation [4b]. Indeed, upon treatment with trimethysilyldiazomethyl lithium, 12 was transformed efficiently into 13.
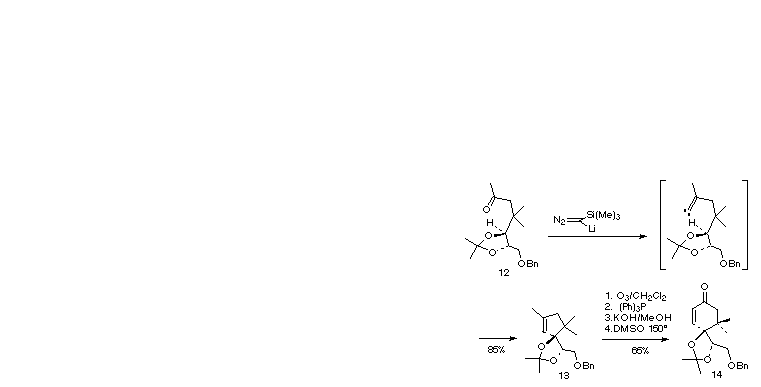
Intramolecular Alkylidene Insertion: Chloromethylenation/ Elimination
The efficiency of the cyclization of 12 to 13 suggested that this could be a good system with which to explore an alternative route1g to alkylidene carbenes, conversion of 12 to the vinyl chloride followed by a-elimination and subsequent C-H insertion to give 13.
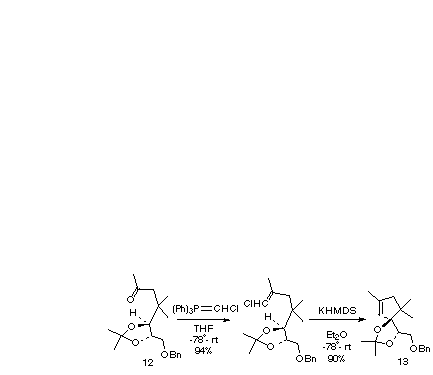
Treatment of the ketone 12 with chloromethylene triphenylphosphorane (prepared by the action of sodium bis(trimethylsilylamide) upon chloromethyltriphenylphosphonium chloride in THF) gives a 50/50 mixture of the E and Z vinyl chlorides. We found that the selection of an appropriate base was critical for eliciting a-elimination and subsequent C-H insertion by the alkylidene carbene. Both sodium hydride and potassium t-butoxide proved to be ineffective. When LDA was used, cyclization was complicated by reduction of the alkylidene carbene to a simple alkene [10]. However, when either sodium or potassium bis(trimethylsilylamide) was used, the exclusive product was 13 in high yield. Thus, in systems for which C-H insertion is facile, chloromethylenation/a-elimination can take the place of treatment with trimethylsilyldiazomethane [11].
Insertion in a Deactivated Methine: Total Synthesis of (+)-Cassiol
With the methodology proven successful in the two extreme cases, we turned our attention to C-H insertion into a deactivated methine, in the context of the total synthesis of the anti-ulcerant (+)-cassiol 1. Our approach to 1 used a convergent strategy. The first task was to create a synthon for the 2, 4-dimethyl-4-hydroxymethyl-2-cyclohexenone ring that forms the core of the molecule. A synthon that would become the 2-ethenyl-1,3-propanediol side chain was also required. For the enone synthon, we employed the vinylogous ester 15. This was to be coupled with the lithio derivative of the side chain synthon, 2, 2-dimethyl-5-(2-bromoethenyl)-1, 3-dioxane, 16, to give (+)-cassiol after hydrolysis. The alcohol 15 was to be elaborated from the cyclopentene 2, the expected product from the reaction of ketone 3 with trimethylsilyldiazomethyllithium.
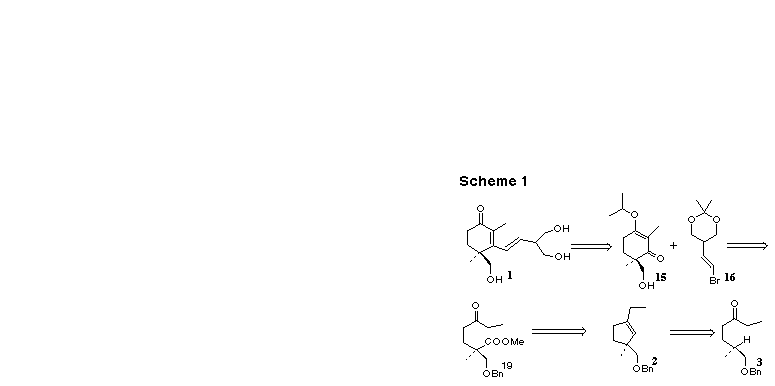
In the event, we prepared enantiomerically pure 3 from (-)-norcitronellol 17 (Scheme 2), available from geraniol following our published procedure [4e]. Benzyl protection of 17 gave an ether 18 which after selective ozonolysis gave the aldehyde. Homologation with ethylmagnesium bromide followed by hydrolysis gave a crude mixture of the diastereomeric secondary alcohols which was oxidized under Swern conditions to give the requisite ketone 3.
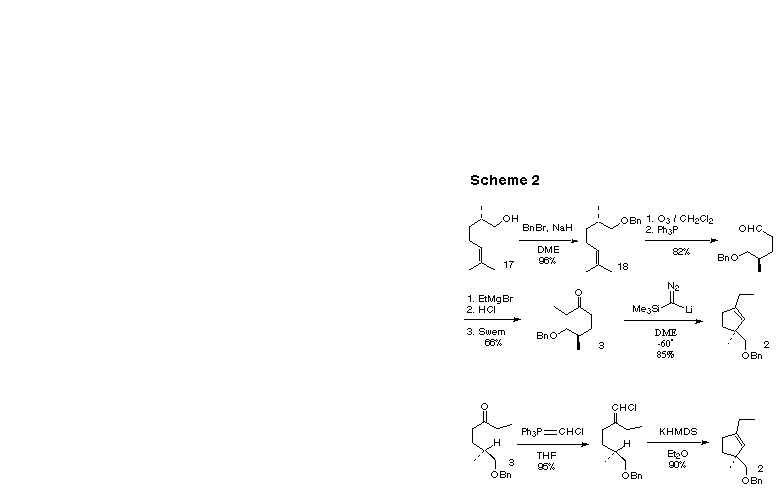
At this point we were ready to attempt cyclization. First we employed our modification of the Ohira cyclization, with the lithium derivative of trimethylsilyldiazomethane [1e, f]. The cyclization was so efficient in this case that we could use n-butyl lithium as the base, with the accompanying hexane solvent.
We then turned our attention to the alternative vinyl chloride cyclization procedure. Treatment of 3 with a THF solution of chloromethylenetriphenylphosphorane gave an excellent yield of a 1:1 mixture of the Z and E vinyl chlorides. Treatment of this mixture with excess potassium bis(trimethylsilyl)amide in ether resulted in the formation of 2 in high yield, underscoring the utility of this procedure for generating alkylidene carbenes.
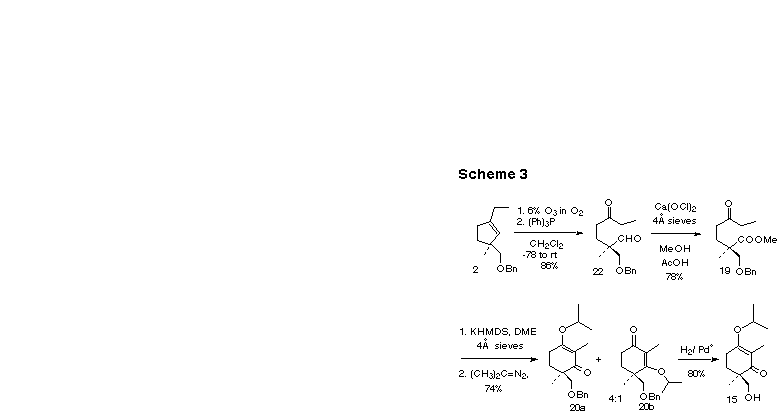
With cyclopentene in hand we carried on through ozonolysis, MacDonald oxidation [12] to the methyl ester 19 and intramolecular Claisen condensation to the desired cyclohexanedione (Scheme 3). We found this vinylogous acid to be unstable, with a room temperature halflife in solution of only a few days, thus complicating its esterification. We solved this problem by effecting esterification with 2-diazopropane [13a] to generate a 4:1 mixture of regioisomers of the vinylogous esters 20a and 20b directly from the crude dione. The regioisomers were readily separable by column chromatography. It is noteworthy that trimethylsilyldiazomethane gave a 1:1 mixture of the two methyl ether regioisomers. Catalytic hydrogenation with 10% Pd-C at room temperature cleanly gave the alcohol 15.
The side chain synthon we developed was 2, 2-dimethyl-5-(2-bromoethenyl)-1, 3-dioxane 16, readily prepared in five steps from commercial diethyl allylmalonate (Scheme 4). First the diester was reduced to the diol [14] with LiAlH4, then the allyl group was isomerized with RhCl3.n H2O and the diol was protected as the acetonide. Ozonolysis of the olefin gave an aldehyde [15] which was condensed with bromomethylenetriphenylphosphorane to form 16 as an approximately equal E/Z mixture.
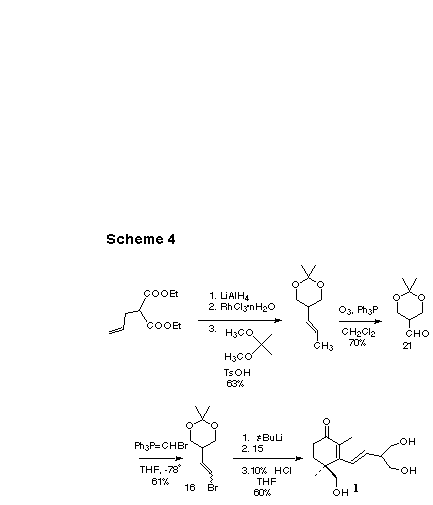
We lithiated 16 using t-butyllithium and added the resulting anion to 15. The intermediate alkoxide was unravelled with aqueous acid to give (+)-cassiol, identical (TLC, 1H, 13C NMR, and MS) with the natural product.
Conclusion
It is apparent that conversion of a ketone to the homologous cyclopentene via the intermediate alkylidene carbene can be achieved efficiently employing trimethylsilyldiazomethyl lithium or by homologation to the chlorovinyl derivative followed by base treatment. We believe that the method outlined here represents a significant step [16] toward reducing to practicality the general cyclopentene synthesis originally adumbrated by Gilbert [1a, b] and Ohira [1c, d].
Acknowledgments: We thank C. Fukaya for providing spectral data of cassioside as isolated in his laboratory. We also thank S. Ohira for his advice. Finally, we are grateful to the NIH (GM 46762) for financial support.
References and Notes
(1) For alkylidene carbene generation using dimethyl(diazomethyl)phosphonate, see (a) Gilbert, J. C.; Giamalva, D. H.; Weersooriya, U. J. Org. Chem 1983, 48, 5251. (b) Gilbert, J. C.; Giamalva, D. H.; Baze, M. E. J. Org. Chem 1985, 50, 2557. For alkylidene carbene generation and cyclization using (trimethylsilyl)diazomethane, see (c) Ohira, S.; Okai, K.; Moritani, T. J. Chem. Soc., Chem. Commun. 1992, 721. (d) Ohira, S.; Sawamoto, T.; Yamato, M. Tetrahedron Lett. 1995, 36, 1537. For our initial reports of the work described in full here, see (e) Taber, D. F.; Meagley, R. P. Tetrahedron Lett. 1994, 35, 7909. (f) Taber, D. F.; Walter, R.; Meagley, R. P. J. Org. Chem. 1994, 59, 6014. (g) Taber, D. F.; Sahli, A.; Yu, H.; Meagley, R. P. J. Org. Chem. 1995, 60, 6571. For leading references to alternative strategies for the generation and subsequent C-H insertion reactions of alkylidene carbenes see (h) Karpf, M; Dreiding, A. S.Helv. Chim. Acta 1979, 62, 852. (i)Ochiai, M.; Uemura, K.; Masaki, Y. J. Am. Chem. Soc. 1993, 115, 2528. (j) Williamson, B. L.; Tykwinski, R. R.; Stang, P. J. J. Am. Chem. Soc. 1994, 116, 93.
(2) For the three previous preparations of (+)-cassiol, see (a) Corey, E. J.; Guzman-Perez, A.; Loh, T. J. Am. Chem. Soc. 1994, 116, 3611. (b) Uno,T.; Watanabe, H.; Mori, K. Tetrahedron 1990, 46, 5563. (c)Takemoto, T.; Fukaya, C.; Yokoyama, K. Tetrahedron Lett. 1989, 30, 723.
(3) (a) Geometries were optimized at the Hartree-Fock level (restricted for the singlet, unrestricted for the triplet) with the 6-31G** basis set. Single point energies were calculated at the MP2 (frozen core) level. Hybridization was analyzed by the Natural Bond Orbital method. (b) Molecular mechanics calculations were carried out using the program Mechanics, implemented on a Tektronix CAChe workstation interfaced with a
Silicon Graphics Indigo workstation. This code is based on the MM2 molecular mechanics code of Allinger, with extensions provided by the CAChe group.
(4) For reviews of diastereoselectivity in Rh-mediated C-H insertion reactions, see (a) Doyle, M. P.; Dyatkin, A. B.; Roos, G. H. P.; Canas, F.; Pierson, D. A.; van Basten, A.; Muler, P.; Polleux, P. J. Am. Chem. Soc. 1994, 116, 4507. (b) Wang, P.; Adams, J. J. Am. Chem. Soc. 1994, 116, 3296. (c) Doyle, M. in Homogeneous Transition Metal Catalysis in Organic Synthesis; Moser, W. R.; Slocum, D. W., Eds.; ACS Advanced Chemistry Series 230; American Chemical Society: Washington DC, 1992; Chapter 30. (d) Taber, D. F. Comprehensive Organic Synthesis, V. 3; Pattenden, G., Ed.; Pergamon Press: Oxford, 1991; p 1045. For more recent references, see (e) Taber, D. F.; You, K. K. J. Am. Chem. Soc. 1995, 117, 5759. (f) Taber, D. F.; You, K. K., Rheingold, A.L. J. Am. Chem. Soc. 1996, 118, 547.
(5) Matsui, M.; Oji, A.; Hiramatsu, K.; Shibata, K.; Muramatsu, H. J. Chem. Soc., Perkin Trans. 2 1992, 201.
(6) (a) Gilbert, J. C.; Blackburn, B. K. J. Org. Chem. 1986, 51, 3656. (b) Gilbert, J. C.; Giamalva, D. H J. Org. Chem. 1992, 557, 4185.
(7) The commercial material also contains impurities that we were unable to remove by distillation.
(8) (a) Wittig, G.; Schlosser, M. Chem. Ber. 1961, 94, 1373. (b) Ketley, A. D.; Berlin, A. J.; Gorman, E.; Fisher, L. P. J. Org. Chem. 1966, 31, 305.
(9) For earlier reports of 1, 5 C-H insertion by alkylidene carbenes generated from vinyl halides, see (a) Erickson, K.L.; Wolinsky, J. J. Am. Chem. Soc. 1965, 87, 1143. (b) Wolinsky, J.; Clark, G. W. J. Org. Chem. 1976, 41, 745. (c) Fisher, R. H.; Baumann, M.; Koebrich, G. Tetrahedron Lett. 1974, 1207.
(10) Creary, X. J. Am. Chem. Soc. 1977, 99, 7632.
(11 )Treatment of the chloromethylene derivative of 9 with KHMDS in diethyl ether resulted in a complex mixture of products, including small amounts of both 10 and 11 , and also the alkyne resulting from 1, 2 methyl shift.
(12) MacDonald, C. E.; Nice, L. E.; Shaw, A. A.; Nestor, N. B. Tetrahedron Lett. 1993, 34, 2741.
(13) (a) For the preparation of 2-diazopropane, see Andrews, S. D.; Day, A. C.; Raymond, P.; Whiting, M. C. Org. Synth. 1970, 50, 27. (b) For the use of 2-diazopropane to esterify vinylogous acids, see Solas, D.; Wolinsky, J. J. Org. Chem., 1983, 48, 670.
(14) Sells, T. B.; Nair, V. Tetrahedron, 1994, 50, 117.
(15) Bates, H. A.; Farina, J.; Tong, M. J. Org. Chem.. 1986, 51, 2637.
(16) Since the inception of our work, several alternative procedures for the generation and cyclization of alklyidene carbenes have been put forward. For leading references, see (a) Tykwinski, R.R.; Stang, P.J.; Persky, N.E. Tetrahedron Lett. 1994, 35, 23. (b) Kunishima, M.; Hikoi, K.; Tani,S.; Kato, A. Tetrahedron Lett. 1994, 35, 7253. (c) Kim, S.; Cho, C. M. Tetrahedron Lett. 1994, 35, 8405. (d) Schildknegt, K.; Bohnstedt, A. C..; Feldman, K. S.; Sambandam, A. J. Am. Chem. Soc. 1995, 117, 7544. (e) Ohira, S.; Sawamoto, T.; Yamato, M. Tetrahedron Lett. 1995, 36, 1537.
(17)Taber, D. F. J. Org. Chem 1982, 47, 1351.
Questions and comments to
Douglass F. Taber