|

|
CERC3
YOUNG
CHEMISTS'
WORKSHOPS
 Friedrich-Alexander-Universität
Erlangen-Nürnberg Friedrich-Alexander-Universität
Erlangen-Nürnberg
|

|
BIOCATALYSIS
2004
March 24 – March 27
in Erlangen, Germany
Institut für Organische Chemie
der Friedrich-Alexander-Universität Erlangen-Nürnberg, Henkestraße 42, D-91054 
CERC3 Young Chemists'
Workshop "Biocatalysis"
March 24 - March
27, 2004
Institut für Organische
Chemie, Henkestr. 42, 91054 Erlangen, Germany

Program
Wednesday,
March 24, 2004
15.00
– 19.00 Registration
19.00 – … Welcome
mixer in the foyer of the great lecture hall of the
Institute of Organic
Chemistry

Thursday, March 25, 2004
09.00 – 09.10 Opening Remarks
Chair: Markus
Reiher
09.10 – 09.45 Mahdi
Abu-Omar (Purdue University, West Lafayette)
“Mechanistic
studies of phenylalanine hydroxylase and a novel example of enzyme self-repair
within the second coordination sphere of catalytic iron”
09.45 – 10.20 Tatyana Polenova (University of Delaware, Newark)
“Solid-State
NMR Spectroscopy for Studies of Geometry and
Electrostatic
Environment in Vanadium Haloperoxidases”
10.20
– 10.55 Roderich Süssmuth (Universität Tübingen)
“Abyssomicins, Novel Inhibitors of the para-Aminobenzoic Acid Biosynthesis
from the Marine Verrucosispora-Strain AB-18-032”
10.55 – 11.20 Coffee break
11.20
– 11.55 Isabel Arends (Delft University of Technology)
“Laccase
Catalysed Aerobic Oxidation of Alcohols”
11.55
– 12.30 Catherine Hemmert (CNRS Toulouse)
“Asymmetric epoxidation with biomimetic catalysts”
12.30
– 13.05 Benjamin Davis (University of Oxford)
“Novel Methods in Carbohydrate-associated Biocatalysis”
13.05 – 14.30 Lunch break
Chair: Norbert Jux
14.30 – 15.05 Markus
Reiher (Universität Bonn)
“The bottom-up approach in theoretical bioinorganic
chemistry”
15.05 – 15.40 Craig
Grapperhaus (University of
Louisville)
“Modeling
Oxygenated Cysteine Thiolates at the Active Site of Iron-Containing Nitrile
Hydratase with a Thiolate/Thioether Donor Set”
15.40 – 16.15 Grit Straganz (Technische Universität
Graz)
“Oxygenases
of the cupin-superfamily - insights into the
mechanism
of the non-heme iron dependent dioxygenase Dke1”
16.15
– 16.45 Coffee break
16.45 – 17.20 Paul Dalby (University College London)
“Expanding the synthetic repertoire of transketolase”
17.20 – 17.55 Michael
Müller (Forschungszentrum Jülich)
“Diversity-oriented
synthesis: new concepts derived from nature”
17.55 – 18.30 Brian
Gibney (Columbia University, New
York)
“Heme Protein
Biocatalysis: Lessons from the Protein Data Bank
and De Novo Heme Protein Design”
19.00 – … Franconian dinner buffet in the
Institute of Organic Chemistry

Friday, March 26, 2004
Chair: Michael
Müller
09.00
– 10.00 Plenary Lecture
Bernhard Lippert (Universität Dortmund)
"Where
Bioinorganic Chemistry Meets Molecular Architecture:
Modelling Metal-Nucleic Acids Interactions"
10.00 – 10.35 Frank Bordusa (MPI für Enzymologie der
Proteinfaltung, Halle)
“Proteases: Old Enzymes with new synthetic properties”
10.35 – 11.00 Coffee break
11.00 – 11.35 Sandrine
Ollagnier de Choudens (Université Joseph Fourier,
Grenoble)
“Biotin synthase: a radical
strategy for sulphur insertion into non-
activated C-H bonds”
11.35 – 12.10 Gideon Grogan (University of York)
“Structure, Mechanism and Application of an unusual Carbon-Carbon
Bond Hydrolase”
12.10 – 12.45 Andrea
Zocchi (Université de Neuchâtel)
“Transgenic
proteins as hosts for enantioselective catalysis”
12.45 – 14.15 Lunch break
Chair: Roderich Süssmuth
14.15 – 14.50 Thibaud
Coradin (Université Pierre et Marie Curie, Paris)
“Biocatalysis in
biomineralization Processes?”
14.50 – 15.25 Michaela Kreiner (University of
Glasgow)
“Protein-coated
micro-crystals: Applications in biocatalysis”
15.25 – 16.00 Cameron Neylon (University of
Southampton)
“Labeling
and immobilizing proteins via intein mediated ligation”
16.00 – 16.30 Coffee break
16.30 – 17.05 Cecilia Tommos (Stockholm University)
“De
Novo Design as a Tool to Investigate Protein Chemistry”
17.05 – 17.40 Virgil
Hélaine (Université Blaise Pascal, Aubiere)
“Directed
evolution on transketolase for obtaining new
monosaccharides”
17.40 – 18.15 Andreas Marx (Universität Konstanz)
“Taming Giants: Insights into
DNA Polymerase Function”
18.15 – Poster
Session in the foyer of the great lecture hall of the Institute of Organic Chemistry,
in between Italian dinner buffet

Saturday, March 27, 2004
Chair: Andreas
Marx
09.00 – 09.35 Rita
Pacheco (Universidade de Lisboa)
“Synthesis of Hydroxamic Acid in TTAB Reversed Micelles
using Amidase from Pseudomonas sp.”
09.35 – 10.10 Erwan
Galardon (Université René Descartes, Paris)
“Modelling the active site of
Nitrile Hydratase: oxidation of (N2S2)-
Fe and (N2S2)-Co
complexes”
10.10 – 10.40 Clotilde Policar (Université Paris IX)
“Mn
complexes as SOD synzymes. Methodology for the evaluation of the SOD-activity:
from anhydrous experiments to transient kinetics in aqueous solution”
10.45 – 11.15 Coffee break
11.15 – 11.50 Gerard Roelfes (University of Groningen)
“From
Enzyme Models to Synthetic Proteins: a Chemist’s Approach to the Study of
Enzymes”
11.50 – 12.25 Jose Palomo (CSIC, Instituto de Catalisis, Madrid)
“Modulation of Chirality of Lipases via immobilization
techniques”
12.25 – 12.30 Closing
remarks
12.30 – 14.00 Lunch buffet in the Institute
of Organic Chemistry
14.00 –
18.30 Excursion to Bamberg
19.00 – Dinner buffet in the Institute
of Organic Chemistry
 CERC3 Young Chemists'
Workshop "Biocatalysis"
CERC3 Young Chemists'
Workshop "Biocatalysis"
March 24 - March 27,
2004
Institut für Organische
Chemie, Henkestr. 42, 91054 Erlangen, Germany

Lecture
Abstracts
(only main authors)
O1 Mahdi Abu-Omar
“Mechanistic
studies of phenylalanine hydroxylase and a novel example of enzyme self-repair
within the second coordination sphere of catalytic iron”
O2 Tatyana Polenova
“Solid-State
NMR Spectroscopy for Studies of Geometry and Electrostatic
Environment
in Vanadium Haloperoxidases”
O3 Roderich Süssmuth
“Abyssomicins, Novel Inhibitors of the para-Aminobenzoic Acid Biosynthesis
from the Marine Verrucosispora-Strain AB-18-032”
O4 Isabel Arends
“Laccase Catalysed
Aerobic Oxidation of Alcohols”
O5 Catherine Hemmert
“Asymmetric
epoxidation with biomimetic catalysts”
O6 Benjamin Davis
“Novel Methods in Carbohydrate-associated Biocatalysis”
O7 Markus Reiher
“The bottom-up approach in theoretical bioinorganic
chemistry”
O8 Craig Grapperhaus
“Modeling
Oxygenated Cysteine Thiolates at the Active Site of Iron-Containing Nitrile
Hydratase with a Thiolate/Thioether Donor Set”
O9 Grit Straganz
“Oxygenases
of the cupin-superfamily - insights into the mechanism of the
non-heme
iron dependent dioxygenase Dke1”
O10 Paul Dalby
“Expanding the synthetic repertoire of transketolase”
O11 Michael Müller
“Diversity-oriented synthesis: new concepts derived
from nature”
O12 Brian Gibney
“Heme Protein Biocatalysis: Lessons
from the Protein Data Bank and De Novo
Heme Protein Design”
O13 Frank Bordusa
“Proteases: Old Enzymes with new synthetic properties”
O14 Sandrine
Ollagnier de Choudens
“Biotin synthase: a radical
strategy for sulphur insertion into non-activated C-H
bonds”
O15 Gideon Grogan
“Structure, Mechanism and Application of an Unusual
Carbon-Carbon bond Hydrolase”
O16 Andrea Zocchi
“Transgenic
proteins as hosts for enantioselective catalysis”
O17 Thibaud Coradin
“Biocatalysis in
biomineralization processes?”
O18 Michaela Kreiner
“Protein-coated
micro-crystals: Applications in biocatalysis”
O19 Cameron Neylon
“Labeling
and immobilizing proteins via intein mediated ligation”
O20 Cecilia Tommos
“De
Novo Design as a Tool to Investigate Protein Chemistry”
O21 Virgil
Hélaine
“Directed
evolution on transketolase for obtaining new monosaccharides”
O22 Andreas Marx
“Taming Giants: Insights into
DNA Polymerase Function”
O23 Rita Pacheco
“Synthesis
of Hydroxamic Acid in TTAB Reversed Micelles using Amidase from Pseudomonas sp.”
O24 Erwan Galardon
“Modelling
the active site of Nitrile Hydratase: oxidation of (N2S2)-Fe
and (N2S2)-Co complexes”
O25 Clotilde Policar
“Mn
complexes as SOD synzymes. Methodology for the evaluation of the SOD-activity:
from anhydrous experiments to transient kinetics in aqueous solution”
O26 Gerard Roelfes
“From
Enzyme Models to Synthetic Proteins: a Chemist’s Approach to the Study of
Enzymes”
O27 Jose Palomo
“Modulation of Chirality of Lipases via immobilization
techniques”
PL Bernhard Lippert
"Where Bioinorganic Chemistry Meets Molecular
Architecture: Modelling
Metal-Nucleic Acids Interactions"
Mechanistic studies of phenylalanine hydroxylase and a novel example of
enzyme self-repair within the second coordination sphere of catalytic iron
Mahdi M. Abu-Omar
Department of Chemistry, Purdue
University, 560 Oval Drive, West Lafayette, IN 47907-2018, U.S.A. Fax:
765-494-0239. E-mail: mabuomar@purdue.edu
Phenylalanine
hydroxylase, a mononuclear non-heme iron enzyme, catalyzes the hydroxylation of
phenylalanine to tyrosine in the presence of oxygen and reduced pterin
cofactor. X-ray structural studies have established the coordination around the
iron metal center and point to significant interactions within the second
coordination sphere. One such interaction involves Tyr325 in human phenylalanine
hydroxylase (hPAH), which forms a hydrogen-bonding network with an aqua ligand
on iron and the pterin cofactor. The full-length tetramer (1-452) and truncated
dimer (117-424) Tyr325Phe hPAH mutant enzymes showed similar kinetics, thermal
stabilities, and oligomerization profiles as their corresponding wild-type
proteins. The possibility of in vivo posttranslational hydroxylation that would
restore the activity of hPAH was examined by mass spectrometry on trypsin
digested full-length (1-452) hPAH Tyr325Phe point mutant. The amino acid tags
obtained by ESI-MS/MS confirmed the presence of a Phe325 in the peptide
corresponding to the doubly charged precursor ion at m/z 916.4 (L A T I F W F T V E F G L C K), and its
hydroxylated counterpart in the peptide corresponding to m/z 924.4 (L A T I F-OH
W F T V E F G L C K) by product ion series comprising the fragments y5-y12.
Furthermore, the point mutation Tyr325Ala resulted in an enzyme that was
totally inactive, and did not display any evidence of hydroxylation. These
results demonstrate the importance of Tyr325 for proper conformation of the
active site, substrate binding, and catalysis. The rescue of the Tyr325Phe
mutant in hPAH via self-hydroxylation presents a novel example of oxidative
repair on the molecular level.
References:
1. A. Volner, J. Zoidakis, M. M.
Abu-Omar, J. Biol. Inorg. Chem., 2003, 8, 121-128.
2. S. D. Kinzie, M. Thevis, K.
Ngo, J. Whitelegge, J. A. Loo, M. M. Abu-Omar, J. Am. Chem. Soc., 2003,
125, 4710-4711.
3. J. Zoidakis, M. Sam, A. Volner,
A. Han, K. Vu, M. M. Abu-Omar, J. Biol. Inorg. Chem., 2004, 9, in press.
Solid-State NMR Spectroscopy for
Studies of Geometry and Electrostatic Environment in Vanadium Haloperoxidases
Tatyana Polenovaa, Neela Pooransingha, Wenlin Huanga, Martin Ebelb, Sven Jantzenb, Dieter Rehderb, Lynn C. Francesconic
a University of
Delaware, Department of Chemistry and Biochemistry, 245 Brown Laboratories,
Newark, DE, USA, Fax: +1 (302)831-6335, e-mail: tpolenov@chem.udel.edu.
b Institut für Anorganische und
Angewandte Chemie, Universität Hamburg, D-20146 Hamburg, Germany; e-mail: rehder@xray.chemie.uni-hamburg.de
c
Department of Chemistry, City University of New York- Hunter College, and the
Graduate Center, 695 Park Avenue, New York, New York 10021, USA; e-mail: lfrances@hunter.cuny.edu
Vanadium
haloperoxidases catalyze a two-electron oxidation of halides to hypohalous acid
in the presence of hydrogen peroxide; the native enzymes require diamagnetic
V(V) for their activity. Vanadium haloperoxidases are the most efficient halide
oxidants known to date, and have attracted significant attention due to their
potential applications in industrial-scale catalytic bioconversions. However,
the mechanism of substrate specificity of these enzymes is poorly understood. The
factors governing substrate specificity remain unclear, especially in the
context of the recent site-directed mutagenesis results. We employ solid-state
NMR spectroscopy to probe the geometry and electrostatic environment of the
vanadium center, as well as the ionization states of the active site amino
acids. In our initial study, we have addressed a series of novel oxovanadium
(V) complexes mimicking the haloperoxidase active site and a series of
vanadium-substituted oxoanionic solids, using a combination of 51V
solid-state Magic Angle Spinning NMR and quantum mechanical calculations with
Density Functional Theory. In the vanadium-substituted ionic oxotungstates, the
fine structure constants are determined by the nature and geometry of counter
cations as well as the vanadium substitution. The NMR spectra provide an
additional measure of sample morphology and positional disorder with respect to
vanadium atoms, which cannot be inferred from the X-ray crystallographic data. The
experimental results revealed that in the haloperoxidase mimics, variations of
the ligand coordination geometry and electronic structure beyond the first
coordination sphere have profound effect on the NMR fine structure constants. Moreover,
for crystallographically characterized compounds density functional theory-
calculations with two different basis sets predict quadrupolar and CSA tensors,
which are in close agreement with the experimental NMR values.
These
findings are of potential importance for understanding differences in
activities due to subtle variations in the active centers of vanadate-dependent
haloperoxidases from different microorganisms.
Solid-state NMR spectroscopy of vanadium haloperoxidases is under way.
This work is
the first step toward correlating the chemical reactivity of the vanadium site
in the vanadium (V) coordination and ionic compounds with their molecular and
electronic structure, which could have further implications in design of
oxovanadium catalysts with tunable properties.
References:
1. Carter JN, Beatty KE, Simpson MT, Butler A
(2002). Reactivity of recombinant and mutant vanadium bromoperoxidase from the
red alga Corallina officinalis, J. Inorg. Biochem.
91, pp. 59-69
2. Tanaka N, Hasan Z, Wever R (2003). Kinetic
characterization of active site mutants Ser402Ala and Phe397His of vanadium
chloroperoxidase from the fungus Curvularia inaequalis, Inorg. Chim.
Acta 256, pp. 288-296
3. Neela Pooransingh, Ekaterina Pomerantseva,
Martin Ebel, Sven Jantzen, Dieter Rehder, Tatyana Polenova (2003). 51V
Solid-State Magic Angle Spinning Spectroscopy and DFT Studies of Oxovanadium
(V) Complexes Mimicking the Active Site of Vanadium Haloperoxidases, Inorg.
Chem. 42, pp. 1256-1266
4. Wenlin Huang, Louis Todaro, Lynn C.
Francesconi, Tatyana Polenova (the 2003). 51V Magic Angle Spinning
NMR Spectroscopy of Six-Coordinate Lindqvist Oxoanions: A Sensitive Probe For
Electronic Environment in Vanadium Containing Polyoxometalates. I. Counterions
Dictate 51V Fine Structure Constants in Polyoxometalate Solids, J.
Am. Chem. Soc. 125, pp.
5928-5938
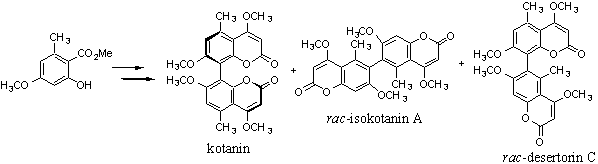
Abyssomicins, Novel
Inhibitors of the para-Aminobenzoic Acid Biosynthesis from the Marine
Verrucosispora-Strain AB-18-032
Bojan Bistera, Julia Riedlingerb,
Andreas Reickeb, Daniel Bischoffa, Hans Zähner, Alan T.
Bull, Louis A. Maldonado, Alan C. Ward, Michael Goodfellow, Hans-Peter Fiedlerb,
Roderich D. Süssmutha
a Universität Tübingen, Institut
für Organische Chemie, Auf der Morgenstelle 18, 72076 Tübingen, Germany, Fax:
+49 (0)7071 295560, e-mail: roderich.suessmuth@uni-tuebingen.de.
b Universität Tübingen,
Mikrobiologisches Institut, Auf der Morgenstelle 28, 72076 Tübingen, Germany,
e-mail: hans-peter.fiedler@uni-tuebingen.de
Organisms of
marine origin are considered as promising source for drug finding. Rare
actinomycetes from the deep sea-plain were screened for inhibitors of the p-aminobenzoate
(pABA) biosynthesis being part of the tetrahydrofolate biosynthesis
pathway. Among the few but prominent synthetic inhibitors of the latter are sulphonamides
and trimethoprim.
A simple agar-diffusion
assay was employed for screening.[1a] Three compounds named
abyssomicins were isolated from Verrucosispora
strain AB 18-032 collected
from a sediment sample in the Japanese Sea. The inhibitory effects of
abyssomicin C as the major metabolite could be depleted upon addition of pABA.
Structure elucidation was performed by ESI-FTICR-MS, 2D-NMR and X-ray
crystallography.[1b] Besides similarities to tetrocarcin-type
antibiotics, the oxabicyclo-partial structure of abyssomicin C resembles the
solution conformation of chorismic acid[2]
as well as synthetic chorismate mutase inhibitors.[3] Furthermore,
the Michael-system (C7-C9) of abyssomicin C lacking in other antibiotically
inactive abyssomicins is supposed to be involved in an irreversible enzyme
trapping-mechanism.
References:
1. a) J. Riedlinger, A. Reicke, B. Krismer, H. Zähner, A.T. Bull,
L.A. Maldonado, M. Goodfellow, B. Bister, D. Bischoff, R.D. Süssmuth, H.-P.
Fiedler, J. Antibiot. 2004, submitted. b) B. Bister, D. Bischoff, M.
Ströbele, J. Riedlinger, A. Reicke, F. Wolter, A.T. Bull, H. Zähner, H.-P.
Fiedler, R.D. Süssmuth, Angew. Chem. 2004, in press.
2. a)
S.D. Copley, J.R. Knowles, J. Am. Chem. Soc. 1987, 109,
5009-5013; b) S.G. Sogo, T.S. Widlanski, J.H. Hoare, C.E. Grinshaw, G.A.
Berchtold, J.R. Knowles, J. Am. Chem. Soc. 1984, 106,
2701-2703.
3. a)
M.C. Kozlowski, N.J. Tom, C.T. Seto, A. Sefler, P.A. Bartlett, J. Am. Chem.
Soc. 1995, 117, 2128-2140; b) P.A. Bartlett, Y. Nakagawa,
C.R. Johnson, S.H. Reich, A. Luis, J. Org. Chem. 1988, 53,
3195-3210.
Laccase Catalysed Aerobic Oxidation of Alcohols
Isabel Arends, Yu-Xin Li, Roger
Sheldon
Delft
University of Technology, Department of Bitechnology, Laboratory for
Biocatalyss and Organic Chemistry, Juliaalaan 136, 2628 BL Delft, Fax: +31
(0)15 2781415, e-mail: i.arends@nw.tudelft.nl.
The selective
oxidation of alcohols is a pivotal reaction in organic synthesis, which is
often hampered by the use of stoichiometric reagents and toxic byproducts. The
development of green, biocatalytic methods for the selective oxidation of
alcohols is therefore an important goal. Copper enzymes are known to catalyse
the aerobic oxidation of alcohols and a well-known example thereof is provided
by galactose oxidase. Another group of copper-dependant oxidases comprises the
laccases (EC 1.10.3.2). These isoenzymes generally contain four copper centers
per protein molecule and catalyse the oxidation of electron rich aromatic
substrates, usually phenols or aromatic amines, via four single electron
oxidation steps concomitant with the four electron reduction of O2
to H2O [1].
In the
delignification of lignocellulose, the action of laccase is enforced by
so-called mediators: low molecular weight electron transfer agents, that
shuttle electrons from the lignin to the enzyme [2]. In our project we use
TEMPO as a mediator to extend the action of laccase towards the oxidation of a
range of alcohols. The following sequence is proposed:
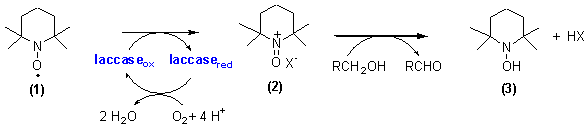
One-electron
oxidation of TEMPO (1) affords the oxoammonium cation (2) which
oxidises the alcohol via a heterolytic patway, giving the carbonyl product and
the hydroxylamine (3). Compound (3) can either be reoxidised by
dioxygen, laccase or the oxoammonium cation (2). Typically the
laccase/TEMPO catalysed aerobic oxidation of alcohols requires high loadings of
TEMPO and laccase, which hampers the commercial application [3]. We have
undertaken a detailed kinetic and mechanistic study in order to develop a more
effective system and to extend the scope to a broad range of alcohol
substrates.
References:
1.
H.P. Call, I. Mucke, J.
Biotechnol. 1997, 53, 163-202; L.
Gianfreda, F. Xu J.M. Bolag, Bioremediation J. 1999, 3,
1-25.
2.
C.B. Eggert, U. Temp and K.E. Eriksson, FEBS Lett., 1996, 391,
144-148.
3.
F. d’Acunzo, P. Baiocco, M. Fabbrini, C. Galli, P. Gentili, Eur. J.
Org. Chem. 2002, 4195-4201.
Asymmetric epoxidation with biomimetic catalysts
Catherine Hemmert, Alexandre
Martinez, Bernard Meunier
Laboratoire de Chimie de Coordination du CNRS, 205 route de
Narbonne, 31077 Toulouse Cedex 4, France, Fax: +33-5-61553003, e-mail:
hemmert@lcc-toulouse.fr.
Optically active epoxides are key intermediates in
organic chemistry because they can undergo stereospecific ring-opening
reactions, giving rise to a wide variety of biologically and pharmaceutically
compounds.[1] There still continues to be great interest in the
design of catalysts for asymmytric alkene epoxidation. The most notable
successes were the chiral manganese salen complexes developed by Jacobsen[2]
et al. and Katsuki[3] et al. at about the same time, which were the
first catalysts able to perform the asymmetric epoxidation of unfunctionalized
olefins in high enantiomeric excess.
In
the present work, we have developed a new strategy to synthesize macrocyclic
chiral manganese salen complexes. The macrocyclisation of the Schiff bases is
expected to increase the stability of the corresponding homogeneous catalysts
for recycling. Moreover, the synthetic strategy allows the modulation of each
building block of the molecules (bulky groups, chiral dimiime and the junction
arm for the macrocyclisation).
References:
1. C. E. Song,
S.-G. Lee, Chem Rev. 2002, 102, 3495-3524.
2. E. N. Jacobsen, M. H. Wu in Comprehensive Asymmetric Catalysis; E.
N. Jacobsen; A. Pfaltz; H. Yamamoto, Eds.; Springer: New York, 1999; Vol. II, pp.
649-677.
3. T. Katsuki in Catalytic Asymmetric Synthesis; 2nd
ed.; I. Ojima, Ed.; Wiley-VCH: New York, 2000;
pp. 287-325.
Novel Methods in Carbohydrate-Associated Biocatalysis
Benjamin
G. Davis
Department
of Chemistry, University of Oxford, Mansfield Road, Oxford OX1 3TA, UK; Fax:
+44 (0)1865 275674; e-mail: Ben.Davis@chem.ox.ac.uk
Sugars
are critical biological markers that modulate the properties of proteins. Our
work studies the interplay of proteins and sugars. This lecture will discuss
recent developments in biocatalysis in our laboratory in two areas: (i)
glycoenzyme synthesis – the use of protein glycosylation to modulate enzyme
function; and (ii) carbohydrate-processing enzyme mechanism – the engineering
and study of glycosidases & glycosyltransferases and the synthesis of
probes of their mechanism.
 (i)
Glycoenzyme synthesis: Glycoproteins occur naturally in mixtures within which
each component has different properties but they are difficult to obtain pure.
To explore the key properties of these glycosylated proteins, there is a
pressing need for methods that will not only allow the preparation of pure
glycosylated proteins, but will also allow the preparation of non-natural
variants for the determination of structure-activity relationships (SARs).[1]
We have described the first examples of an answer to this problem which allows
site- and sugar- specific glycosylation of proteins using a novel combined
site-directed mutagenesis (SDM) and chemical modification strategy.[2] This has
allowed the effects of protein glycosylation to be explored precisely for the
first time.[3] Precisely glycosylated enzymes can be used in • preparative
biocatalysis[4] • drug delivery[5] and • selective protein degradation[6]. A
new class of glycoconjugate, the glycodendriprotein,
has also been developed[7] that acts as a powerful nanomolar inhibitor of
bacterial interactions. Furthermore, 2nd generation glycosylating
reagents[8] allow the incorporation of the largest known carbohydrates in site
selective glycosylations yet require only very small amounts of sugar.
(i)
Glycoenzyme synthesis: Glycoproteins occur naturally in mixtures within which
each component has different properties but they are difficult to obtain pure.
To explore the key properties of these glycosylated proteins, there is a
pressing need for methods that will not only allow the preparation of pure
glycosylated proteins, but will also allow the preparation of non-natural
variants for the determination of structure-activity relationships (SARs).[1]
We have described the first examples of an answer to this problem which allows
site- and sugar- specific glycosylation of proteins using a novel combined
site-directed mutagenesis (SDM) and chemical modification strategy.[2] This has
allowed the effects of protein glycosylation to be explored precisely for the
first time.[3] Precisely glycosylated enzymes can be used in • preparative
biocatalysis[4] • drug delivery[5] and • selective protein degradation[6]. A
new class of glycoconjugate, the glycodendriprotein,
has also been developed[7] that acts as a powerful nanomolar inhibitor of
bacterial interactions. Furthermore, 2nd generation glycosylating
reagents[8] allow the incorporation of the largest known carbohydrates in site
selective glycosylations yet require only very small amounts of sugar.
(ii)
Carbohydrate-Processing Enzymes: Glycosidases are powerful tools for use in
glycoside synthesis and potential targets for therapeutic intervention. Many
elegant studies have made significant advances towards understanding the
reactive catalytic mechanisms of these enzymes. However, the mechanism by which
substrate specificity is determined is still largely unclear. We have begun to
explore the underlying basis of substrate selectivity through mutagenesis to
tailor the substrate tolerance of glycosidase catalysts and enhance their
synthetic utility.[9] In addition, we have developed methods[10] for the ready
construction of arrays of inhibitors as probes of carbohydrate-processing
enzymes and which have allowed the identification of novel inhibitors.[10]
These include novel stereodynamic aza-sugar strategies that have allowed the
first synthesis of the naturally occurring hydrophobically-modified aza sugar,
Adenophorine.[12] In addition, through the use of a novel high throughput mass
spectrometric screening system we have begun to evaluate the specificity and
activity of the entire 107 strong A thaliana glycosyltransferase family
GT-1. Initial results reveal some unusual activities and highlight some
striking functional similarities with some bacterial family GT-1 enzymes.
References:
[1] B.G.
Davis, Chem. Rev. 2002, 102, 579-601.
[2] B.G.
Davis, M.A.T. Maughan, M.P. Green, A. Ullmann, J.B. Jones, Tetrahedron: Asymm. 2000, 11, 245-262; U.S. Patent Application
US 09/347,029, PCT Intl Application
WO 0001712; Chem. Abs., 132, 89792: Filed July 2, 1998;
[3] B.G. Davis, R.C. Lloyd and
J.B. Jones, Bioorg. Med. Chem. 2000, 8, (7), 1527-1535.
[4] K. Matsumoto, B.G. Davis,
J.B. Jones, Chem. Commun. 2001, 903-904; K. Matsumoto, B.G.
Davis, J.B. Jones, Chem. Eur. J. 2002, 4129-4137.
[5] M.A. Robinson and B.G.
Davis, Curr. Opin. Drug Discovery
Development 2002, 5, (2), 279-288.
[6] B.G Davis et al, ChemBioChem 2003, 4, 533-537.
[7] B.G. Davis, Chem. Commun. 2001, 351-352; P.M. Rendle, A.P. Seger, J. Rodrigues,
N.J. Oldham, R.R. Bott, J.B. Jones, M.M. Cowan and B.G. Davis, J. Am. Chem. Soc., 2004, in press.
[8] D.P.
Gamblin, P. Garnier, S.J. Ward, N.J. Oldham, A.J. Fairbanks, B.G. Davis, Org. Biomol. Chem., 2003, 1, 3642-3644; D.P. Gamblin, P. Garnier, S. van
Kasteren, N.J. Oldham, A.J. Fairbanks and B.G. Davis, Angew. Chem. 2004, 116,
(7) 845-851; Angew. Chem. Int. Ed. 2004, 43, (7) 827-833.
[9] K. Corbett, A.P.
Fordham-Skelton, J.A. Gatehouse, B.G. Davis, FEBS Lett. 2001, 509, 355-360.
[9] B.G. Davis, M.A.T. Maughan,
T.M. Chapman, R. Villard, S. Courtney, Org.
Lett., 2002, 4, 103-106.
[10] T.M. Chapman, S. Courtney, P.
Hay, B.G. Davis, Chem. Eur. J., 2003, 9, 3397-3414.
[11] M.A.T. Maughan, I.G. Davies,
T.D.W. Claridge, S. Courtney, P. Hay, B.G. Davis, Angew. Chem. Intl Ed. 2003,
42, 3788.
The bottom-up approach in theoretical bioinorganic catalysis
Markus Reiher
Universität Bonn, Lehrstuhl für Theoretische Chemie,
Wegelerstr. 42, D-53115 Bonn, Germany, Fax: +49 (0)228 739064,
e-mail: reiher@thch.uni-bonn.de
For arriving at a detailed
microscopic understanding of reactions taking place in metallo-enzymes,
theoretical methods are urgently needed in order to supplement experimental
data. Prominent examples, where experimental and theoretical methods compete
and yield complementary results, are the yet unknown mechanisms of nitrogenase
and NiFe hydro-genase.
While theoretical methods
allow for a rigorous control of errors (at least in principle), methods of density
functional theory (DFT) still suffer of serious failures. Because of the fact
that the activity of metalloenzymes is largely determined by the transition
metal atom or cluster in the active center, it is inevitable to establish
accurate and efficient quantum chemical methods for reliable calculations of
such electronic structures. We set out to study failures and benefits of
current DFT methods on experimentally well characterized biomimetic model
systems and models of active sites in metalloenzymes in order to improve on the
presently available methodologies.
The talk starts with a
discussion of structures and energetics of Sellmann-type nitrogenase model
complexes, which will reveal serious deficiencies of standard density
functionals with respect to the calculation of spin state energetics (with
consequences for the calculation of reaction energies).
Furthermore, understanding
electronic structures requires tools for qualitative analyses of electronic
wavefunctions. Here, we will discuss a novel semi-quantitative method for the
fast estimation of intramolecular hydrogen bond energies as well as an
implementation of Davidson’s local spin definition applied to the FeMo-cofactor
of nitrogenase.
Next, the calculation of
molecular properties in a rigorous quantum mechanical framework requires smart
algorithms, which do not calculate all but only the chemically relevant
properties. The talk demonstrates how this can be achieved for molecular
vibrations, for which we have established the so-called Mode-Tracking
methodology.
References:
1. Please, visit our web pages at www.thch.uni-bonn.de/tc/groups/reiher
for detailed information and references
Modeling Oxygenated Cysteine Thiolates at the Active Site of Iron-Containing
Nitrile Hydratase with a Thiolate/Thioether Donor Set
Craig A. Grapperhausa, Ming Lia, Apurba
K. Patraa, Selma Poturovica, Pawel M. Kozlowskia, Marek Z. Zgierskib, Mark S. Mashutaa
aUniversity
of Louisville, Department of Chemistry, Louisville, Kentucky, USA, 40292,
e-mail: grapperhaus@louisville.edu
bSteacie Institute for Molecular
Science, National Research Council of Canada, Ottawa, Ontario, Canada, K1A OR6
Nitrile hydratase
(NHase), which catalyzes the hydration of nitriles to amides, is an intriguing
enzyme not only based on the utility of its function, but also the uniqueness
of its active site.1 The enzyme incorporates iron,
normally associated with redox active enzymes, in a non-redox role. Secondly, the N2S3
donor environment about the iron contains two modified cysteine residues
resulting in a mixed thiolato (RS-), sulfenato (RS(O)-),
sulfinato (RSO2-) donor set. Such sulfur oxygenation is
unprecedented in biological systems and its consequences remain undetermined. A
series of iron complexes based on the pentadentate ligand 4,7-bis(2’-methyl-2’-mercaptopropyl)-1-thia-4,7-diazacyclononane),
(bmmp-TASN)2-, have been synthesized and characterized as models of
iron-containing NHase.2 The ligand binds
iron(III) with a single, variable additional ligand, X (X = Cl, NO, CN, OFeL).
As demonstrated by EPR and NMR, the nature of X, and not the presence of the
two π-donating thiolate
donors, determines the spin-state of the complex. Comparison of the metric data
within the series highlights a key difference between high-spin and low-spin
iron-thiolate bonding.2,3 Whereas the
iron-thiolate and iron-thioether bond distances are quite different for
high-spin iron(III) they are indistinguishable for low-spin iron. The relevance
of the spectroscopic and structural results to nitrile hydratase is described
and the similarities and differences of thiolates, thioethers, and S-oxygenates
as donors to low-spin iron are presented. Density functional theory (DFT)
calculations of the iron-nitrosyl complex are consistent with our description
of iron-sulfur bonding in this system and reveals the HOMO region is dominated
by Fe-S bonding.
References:
1. Shigehiro, S.; Nakasako, M.;
Dohmae, N.; Tsujimura, M.; Tokoi, K.; Odaka, M.; Yohda, M.; Kamiya, N.; Endo,
I., Nat. Struct. Biol. 1998, 5, 347-351.
2. Grapperhaus, C. A.; Li, M.; Patra, A. K.; Poturovic, S.;
Kozlowski, P. M.; Zgierski, M. Z.; Mashuta, M. S., Inorg. Chem. 2003, 42, 4382-4388.
3. Grapperhaus, C. A.; Patra, A. K.; Mashuta, M. S., Inorg. Chem. 2002, 41, 1039-1041.
Oxygenases of the cupin-superfamily
- insights into the mechanism of the non-heme iron dependent dioxygenase Dke1
Grit Straganza, Bernd Nidetzkyb
a Technische Universität Graz,
Institute of Biotechnology and Biochemical Engineering, Petersgasse 12, 8010
Graz, Austria, Fax: +43 (0)316 873-8434, e-mail: grit.straganz@TUGraz.at.
b Technische Universität Graz,
Institute of Biotechnology and Biochemical Engineering, Petersgasse 12, 8010
Graz, Austria, Fax: +43 (0)316 873-8434, e-mail: bernd.nidetzky@TUGraz.at.
The cleavage of C-C bonds via molecular oxygen by
non-heme metal dependent dioxygenases plays an essential role in numerous
biochemical pathways. Although ubiquitous in nature, this reaction class does
not have a direct chemical counterpart. The requirements for the active site of
a natural or biomimetic metal-complex
to trigger oxygenative C-C bond cleavage are under active investigation.
While especially the C-C bond cleavage of intradiol and extradiol cleaving
catechol dioxygenases have been subject of intense study, reports on oxygenases
of the cupin family are limited (1). The latter are very diverse regarding
their metal cofactor as well as their substrate acceptance. The diketone
cleaving dioxygenase Dke1 [EC 1.13.11.50] is one exponent of the cupin-super
family (2,3). Its relaxed substrate spectrum towards basic b-dicarbonyl structures (4) offers interesting new
possibilities for the investigation of the enzyme mechanism – giving us the
possibility to directly study electronic effects on various steps of the enzme
mechanism, such as the oxygenative C-C bond cleavage. Our findings offer
intriguing insights into the principle nature of oxygen activation and C-C bond
cleavage in non-heme metal dependent dioxygenases.
References:
1. Jim M.
Dunwell, Sawsan Khuri, Paul J. Gane, Microbiology and Molecular Biology
Reviews, 2000, 64, 153-179.
2. Gudrun R. Stranzl, Strukturuntersuchungen
an Enzymen im Kristall und in Loesung, Dissertation, Universität Graz, 2002.
3. Grit D. Straganz, Lothar Brecker, Hns-Jörg Weber, Walter Steiner,
Douglas W. Ribbons, Biochem. Biophys.
Res. Commun. 2002, 297, 232-236.
4. Grit D. Straganz, Anton Glieder, Lothar Brecker, Douglas W.
Ribbons, Walter Steiner, Biochem J. 2003, 369, 573-581.
Expanding the synthetic
repertoire of transketolase
Paul A. Dalby
Dept Biochemical Engineering, University College London, Torrington Place, London, WC1E 7JE,
UK, Tel: 020 7679 2962,
e-mail: p.dalby@ucl.ac.uk.
Transketolase
catalyses the transfer of a two-carbon ketol unit from xylulose 5-phosphate to
an aldehyde acceptor such as erythrose 4-phosphate, producing a new C-C bond
and chiral centre with high enantioselectivity. An alternative ketol donor,
beta-hydroxypyruvate, can be used to ensure that the reaction is irreversible,
producing CO2 as a byproduct, and hence cost effective at large
scale.
To
widen the appeal of using enzymes as catalysts for organic synthesis it is desirable
to improve their properties in terms of catalyst stability in non-physiological
conditions and acceptance of a broader range of substrates. Our efforts to address both of these issues
will be presented.
Firstly,
we aim to improve enzyme stability by directed evolution and thus require a
screening method that directly measures enzyme stability. A screening approach will be presented in
which enzyme unfolding can be measured directly in an automatable microwell
system, thus avoiding the need to use indirect screens such as
thermoinactivation assays.
Secondly,
we are exploring the use of bioinformatics combined with genetic engineering to
obtain a broad-range transketolase that could be used more generally in organic
synthesis, but also as a valuable starting point for directed evolution in the
development of large-scale biocatalytic routes. The methodology for identifying potential broad-range enzymes
will be presented and the results of preliminary kinetic characterisations will
be rationalised in terms of structural considerations.
References:
1.
Dalby, P. A. (2003) Curr. Opin.
Struct. Biol. 13 (4), 500-505. Optimising enzyme function by directed
evolution.
2.
Lye, G. J., Ayazi-Shamlou, P., Baganz, F., Dalby, P. A., and Woodley, J. M. (2003) Trends Biotech. 21 (1),
29-37. Accelerated design of bioconversion processes using automated microscale
processing techniques.
3. Lye, G. J., Dalby,
P. A., and Woodley, J. M. (2002) Org. Process
Res. Dev. 6 (4), 434-440. Better
biocatalytic processes faster: New tools for the implementation of biocatalysis
in organic synthesis.
Diversity-oriented
synthesis: ‘new’ concepts derived from nature
Michael Müller
Institut
für Biotechnologie 2, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany,
Fax: +49 (0)2461 913870, e-mail: mi.mueller@fz-juelich.de.
Target-oriented synthesis has
been the major task of synthetic organic chemistry. Diversity-oriented
synthesis became a major point of interest within the last decade. Aspects of
both strategies are used by nature for the biosynthesis of highly specific
molecules as well as a broad diversity of many compounds with unknown
biological function.1 In our own work we utilize
diversity-oriented aspects according to biosynthesis in (target-oriented)
organic synthesis. This has been applied e.g. in the synthesis of regioisomeric
natural products,2 for the development of different ThDP-dependent enzyme-catalyzed
reactions,3 and for the microbial production of ‘unnatural’
metabolites.4
Natural product synthesis:
Metabolic engineering:
References:
1. R. D. Firn, C. G. Jones, Nat. Prod. Rep. 2003, 20, 382-391.
2. a) D. Drochner, W. Hüttel, M. Nieger, M. Müller, Angew. Chem. 2003, 115, 961-963; Angew. Chem. Int. Ed. 2003,
42, 931-933; b) W. Hüttel, M.
Nieger, M. Müller, Synthesis 2003, 1803-1808.
3. M. Pohl, B. Lingen, M. Müller, Chem.
Eur. J. 2002, 8, 5288-5295.
4. a) Angew. Chem. 2001,
113, 578-581; Angew. Chem. Int. Ed. 2001, 40, 555-557; b) D. Franke, G. A. Sprenger, M. Müller, CHEMBIOCHEM 2003, 4, 775-777.
Heme Protein Biocatalysis: Lessons from the Protein Data Bank and De
Novo Heme Protein Design
Brian Gibney, Jinyou
Zhuang, Charles J. Reedy, Jennifer H. Amoroso
Columbia University, Department of
Chemistry, 3000 Broadway, MC 3121, New York, NY 10027 Fax: (212) 932 1289,
email: brg@chem.columbia.edu
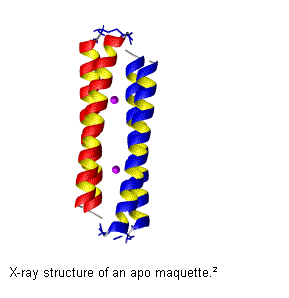 Our approach to
the study of metalloproteins critical to biocatalysis is to engineer and
fabricate peptide structures that incorporate metal cofactors toward the goal
of generating molecular maquettes,
simplified functional versions of complex native enzymes. Herein, we have
analyzed all structurally characterized natural heme proteins1 to
design novel synthetic heme protein maquettes for the investigation of the fundamental
design principles natural heme proteins. The designed four helix bundle
protein, shown at right, is elaborated with histidine ligands to bind heme,
Fe(protoporphyrin IX). Using heme protein maquettes, we have delineated the
environmental factors which alter the heme reduction potential, a fundamental
chemical property of natural cytochromes. The type of porphyrin macrocycle, the
local electrostatic environment, the burial of the heme and the influence of pH
all contribute to the modulation of the heme reduction potential. In designed
heme proteins, we can modulate the heme reduction potential by 435 mV - nearly half the range observed for natural
heme proteins. Evaluation of the Fe(III) and Fe(III) heme binding constants
and the resultant electrochemistry provides insight into the absolute
(de)stabilization of these states by the protein environment.3 We have
expanded the repertoire of ligands available for heme protein design by using
nonnatural amino acids containing pyridine, triazole and tetrazole sidechains.4 Altering
the axial ligands leads to significant changes in heme spectroscopy (electron
paramagnetic resonance, magnetic circular dichroism, resonance Raman),
reduction potential (+286 mV) and Fe(III) and Fe(II) stability constants
(150,000-fold). These results will be compared with those from the set of
structurally characterized natural heme proteins.
Our approach to
the study of metalloproteins critical to biocatalysis is to engineer and
fabricate peptide structures that incorporate metal cofactors toward the goal
of generating molecular maquettes,
simplified functional versions of complex native enzymes. Herein, we have
analyzed all structurally characterized natural heme proteins1 to
design novel synthetic heme protein maquettes for the investigation of the fundamental
design principles natural heme proteins. The designed four helix bundle
protein, shown at right, is elaborated with histidine ligands to bind heme,
Fe(protoporphyrin IX). Using heme protein maquettes, we have delineated the
environmental factors which alter the heme reduction potential, a fundamental
chemical property of natural cytochromes. The type of porphyrin macrocycle, the
local electrostatic environment, the burial of the heme and the influence of pH
all contribute to the modulation of the heme reduction potential. In designed
heme proteins, we can modulate the heme reduction potential by 435 mV - nearly half the range observed for natural
heme proteins. Evaluation of the Fe(III) and Fe(III) heme binding constants
and the resultant electrochemistry provides insight into the absolute
(de)stabilization of these states by the protein environment.3 We have
expanded the repertoire of ligands available for heme protein design by using
nonnatural amino acids containing pyridine, triazole and tetrazole sidechains.4 Altering
the axial ligands leads to significant changes in heme spectroscopy (electron
paramagnetic resonance, magnetic circular dichroism, resonance Raman),
reduction potential (+286 mV) and Fe(III) and Fe(II) stability constants
(150,000-fold). These results will be compared with those from the set of
structurally characterized natural heme proteins.
References:
1. Reedy, C.J.; Gibney, B.R. Chem. Rev., 2004, 101, 617-649.
2. Huang, S.S.; Gibney, B.R.;
Stayrook, S.E.; Dutton, P.L.; Lewis, M J.
Mol. Biol., 2003, 326, 1219-1225.
3. Reedy, C.J.; Kennedy, M.L.;
Gibney, B.R. Chem. Commun., 2003, 570-571.
4. Privett, H.K.; Reedy, C.J.;
Kennedy, M.L.; Gibney, B.R. J. Am. Chem.
Soc., 2002, 124, 6828-6829.
Proteases: Old enzymes with new synthetic properties
Nicole Wehofskya, Kathrin
Ralla, Angela Pöhlmanna, Andreas Pecha, Stephanie
Schmidta, Sven Klußmannb, Hidenobu Komedac,
Yasuhisa Asanoc, Frank Bordusaa
a Max-Planck Research Unit for
Enzymology of Protein Folding, Weinbergweg 22, D-06120 Halle/Saale, Germany,
Fax: +49 (0)345 5511972, e-mail: bordusa@enzyme-halle.mpg.de.
b NOXXON
Pharma AG, Max-Dohrn-Strasse 8-10, D-10589 Berlin, Germany
c
Biotechnology
Research Center, Toyama Prefectural University, Toyama 939-0398, Japan
Forced by
successful enzyme, medium and substrate engineering methods, proteases have
gained in importance as regio- and stereospecific catalysts in organic
synthesis.[1] Especially for applications that are based on their
native hydrolysis activity, such as regiospecific ester hydrolysis or the
kinetic resolution of racemates, proteases are now generally recognized as
normal bench reagents. In principle, these engineering methods allow also for
the reduction of competitive acyl donor hydrolysis, the alteration of the
enzyme specificity and the minimization of undesired proteolytic cleavages,
which are the main drawbacks when proteases are used as catalysts for reverse
proteolysis. However, even the most impressive examples published so far are
handicapped by the intrinsic drawbacks of the protease catalyst that are
mainly: i) the restricted specificities and selectivities of the available
proteases and ii) the permanent risk of proteolytic side reactions of both the
starting compounds and the products formed. Summarizing these characteristics,
proteases appear to be far away from being perfect catalysts for universal and
flexible peptide synthesis. Inevitably, this conclusion holds true for
classical approaches, but is wrong when considered as a general rule. In fact,
the combined use of substrate mimetics,[2] genetically optimized
peptide ligases[1] and solid phase peptide synthesis approaches[1]
have broaden the synthetic scope and flexibility of the enzymatic method. This
allows for the coupling and modification of cleavage-sensitive peptide
fragments and the selective labeling of polypeptides at the N-terminus in an irreversible manner.
Besides the coupling of coded amino acid moieties a broad spectrum of noncoded
once, such as D-amino acids,[3]
carbohydrate moieties[4] or nonpeptidic carboxylic acids,[5]
undergo the coupling approach and further broaden its scope for organic
synthesis. Selected original examples including the synthesis of longer
polypeptides, peptide isosteres, structural diverse N-linked peptidoglycans, and the selective coupling of non-amino
acid derived carboxylic acids to peptides and proteins will be presented.
Particular attention will be paid to the synthetic utility of this powerful
chemoenzymatic approach and to its unique degree of flexibility.
References:
1. F. Bordusa, Chem. Rev. 2002, 102, 4817-4867.
2. F.
Bordusa, D. Ullmann, C. Elsner, H.-D. Jakubke, Angew. Chem. Int. Ed. 1997, 36, 2473-2475.
3. N. Wehofsky, R. Löser, A.
Buchynskyy, P. Welzel, F. Bordusa, Angew.
Chem. Int. Ed. 2002, 41, 2735-2738.
4. N. Wehofsky, S. Thust, J.
Burmeister, S. Klussmann, F. Bordusa, Angew.
Chem. Int. Ed. 2003, 701-704.
5. R. Günther, F. Bordusa, Chem. Eur. J. 2000, 6,
463-467.
Biotin synthase: a radical
strategy for sulphur insertion into non activated C-H bonds
S. Ollagnier-de Choudens, J. Rubach, E. Mulliez,
M. Fontecave
Laboratoire de Chimie et Biochimie des
Centres Rédox Biologiques, DBMS-CB, CEA/CNRS/Université Joseph Fourier, UMR
5047, 17 Avenue des Martyrs, 38054 Grenoble Cedex 09, France. Tel: 00(33)438789115, Fax:
00(33)438789124, email: sollagnier@cea.fr
Biotin synthase
catalyzes the last step of the biotin biosynthesis pathway. The reaction
consists in the introduction of a sulfur atom into dethiobiotin, thus requiring
activation of C-H bonds. Radical activation of the dethiobiotin substrate is
performed by the association of two biotin synthase cofactors, the iron-sulfur
cluster and S-adenosylmethionine. Recently we have identified a new enzymatic
activity of biotin synthase, a cysteine desulfurase activity, which may provide
the sulfur introduced into biotin. This activity is dependent on
pyridoxal-5-phosphate and proceeds through a protein bound persulfide. By
site-directed mutagenesis experiments two conserved cysteines residues were
shown to be critical for this activity and are good candidates as a site for
persulfide. A new mechanism for sulfur insertion into dethiobiotin, in which
persulfide play a key role, is proposed.
Structure, Mechanism and Application of an Unusual Carbon-Carbon Bond
Hydrolyase
Philip M. Leonard, Jean L.
Whittingham, Gideon Grogan
York
Structural Biology Laboratory, University of York, Heslington, York YO10 5YW
U.K.
Fax: +44
1904 328266, e-mail: grogan@ysbl.york.ac.uk.
6-oxocamphor
hydrolyase (OCHL) is a cofactor independent enzyme that catalyses the
desymmetrisation of bicyclic β-diketones
to yield cyclic keto acids of high optical purity via an enzymatic retro-Claisen
reaction (Figure) [1,2].
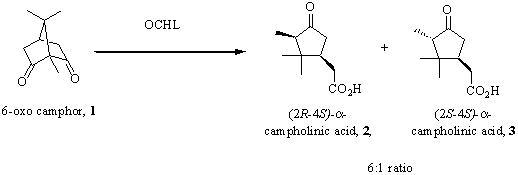
The structure of
the enzyme has been solved to 2Å resolution [3], the active site located, and a
number of site-directed mutants prepared in an effort to shed light on the
mechanism of the enzyme. The low kcat plus low KM
mutant His122Ala crystallised in complex with the minor diastereomer of the
natural product, (2S, 4S)-a-campholinic acid.
This ligand complex reveals many of the molecular determinants of prochiral
selectivity in the enzyme, provides clues to mechanism and also provides
suggestions for engineering isozymes of opposite selectivity.
References:
1. G. Grogan, J. Graf, A. Jones, S. Parsons, N.J. Turner and S.L.
Flitsch Angew. Chem. Intl. Ed. Engl., 2001, 40, 1111-1114.
2. G. Grogan, G.A. Roberts, D. Bougioukou, N.J. Turner and S.L.
Flitsch J. Biol. Chem., 2002, 275, 12565-12572.
3. J.L. Whittingham, J.P. Turkenburg, C.S. Verma, M. A. Walsh and G.
Grogan J. Biol. Chem., 2003, 278, 1744 –1750.
Transgenic
proteins as hosts for enantioselective catalysis
Andrea Zocchi, Nicolas Humbert, Jérôme Collot, Julieta Gradinaru,
Myriem Skander, Andreas Loosli, Gérard Klein, Thomas R. Ward
Institute of
Chemistry, University of Neuchâtel, rue E. Argand 11, CH-2000 Neuchâtel
Fax: +41 (0)32 7182511,
e-mail: andrea.zocchi@unine.ch, thomas.ward@unine.ch
Biochemical and physiological processes utilize
preferentially one enantiomer over its mirror-image. Infact, small mirror-image
impurities may cause severe pharmacological and toxicological side effects.
Thus, a major goal in chemistry is to generate enantiomerically pure compounds.
We produce artificial metalloenzymes introducing stereoselectivity in catalyses
that normally generate a racemate. The principle of our metalloenzymes lies on
the incorporation of active metal catalyst precursors in a well-defined chiral
protein environment, by the biotin-(strept)avidin technology. This technology
ensures the precise localization of a biotinylated moiety without the need of a
chemical coupling step with the host protein.
Expression systems. Avidin is a homotetrameric
glycoprotein found in avian, reptilian and amphibian egg white. Each monomer
can bind a (+)-biotin (vitamin H) molecule with an extraordinary affinity (Ka=
~1014 M-1). We have overexpressed an artificial avidin
gene in Pichia pastoris. The recombinant protein is secreted in the
extracellular medium with a maximal yield of 400 mg/L. The purification only requires
the basification of the culture medium prior to affinity chromatography.
Streptavidin is the prokaryotic counterpart of avidin, with the
same tridimensional eight-stranded b-barrel homotetrameric
structure, but only 32% protein sequence homology. It is produced by Streptomyces
avidinii. Each of subunit can bind a (+)-biotin molecule with almost the
same affinity as avidin. We have overexpressed a streptavidin gene in E.
coli. The recombinant protein is targeted into the cytoplasm. The
multi-step purification involves lysis of the cells, enzymatic digestion of
nucleic acids and a denaturing-renaturing step prior to affinity
chromatography. In these conditions, we obtain up to 160 mg of streptavidin per
L of culture. Streptavidin is also overexpressed in P. pastoris with the
same yield as avidin.
Mutagenesis. Site directed mutagenesis and Site
saturation mutagenesis are utilized for modification of the loops
connecting b-sheets around the biotin-binding pocket. In
most cases, the original amino acid was exchanged with a glycine, to confer
more flexibility to the loop. Synthesis of an artificial gene is
utilized to optimize the expression of streptavidin or avidin in a given
expression system and to create chimerical proteins. These three methods are used
to elucidate the factors responsible for enantioselection.
Main results. Streptavidin proved to be a better chiral inducer
than avidin in the vast majority of experiments. For example, the best
enantiomeric excess (ee) for the reduction of acetamidoacrylic acid with
a mutant streptavidin as host protein affords acetamidoalanine with 96% ee
(R). The best ee with mutant avidin for the same substrate was only 77%
(S). Preliminary results also indicate that a chimerical avidin bearing one
loop of streptavidin behaves more like streptavidin.
References:
1. J.
Collot et al. J. Am. Chem.
Soc. 2003, 125, 9030-9031.
2. T. R. Ward
et al. Chimia. 2003, 57, 586-588.
3. A. Zocchi
et al. Chimia. 2003, 57, 589-592.
4. A. Zocchi
et al. Prot. Expr. Pur. 2003, 32,
167-174.
5. J. Collot et al. J. Am. Chem. Soc. 2004, submitted.
Biocatalysis in Biomineralization Processes?
Thibaud Coradin, Jacques
Livage
Laboratoire de Chimie de la Matière Condensée, CNRS-UMR
7574, Université Pierre et Marie Curie, 4 place Jussieu, 75252 Paris cedex 05,
France, Fax: +33 (0)144274769, e-mail: coradin@ccr.jussieu.fr.
When considering the role of inorganic species in
biochemical processes, only isolated ions or metalloprotein active sites are
often taken into account. However, biominerals, i.e. solid phases deposited by
living organisms, are widespread examples of inorganic species present in a
condensed state. The question arises whether some specific biomolecules are
involved in the formation of these biogenic materials. This presentation will
try to give an overview of the actual knowledge in this field, especially
focusing on silica-based biominerals.1
In the case of calcium salts like phosphate in bones
or carbonate in mollusk shells, some proteins and poly-saccharides have been
shown to favour precipitation as well as to control the morphology and crystal
structure of the deposited solid. However, no real catalytic activity of these
biomolecules have been demonstrated.
In contrast, for silica present in several plants,
algae or sponges, the possibility for proteins to interact specifically with
silica molecular precursors has been suggested. Some of these biomolecules were
isolated and their activity towards silica formation was studied. In parallel,
model systems were used to get a better understanding of the interactions that
may arise between proteins and silica precursors.
These results suggest that although different
biomolecules can activate silica formation, a true catalytic process has not
yet been unambigously identified at this time. This can be partially attributed
to the fact that the silica precursors used by the organisms, as well as their
reactivity within the cells, are still poorly known. Nevertheless, in addition
to their fundamental implications, these studies have already been fruitful in
designing new silica-based “bio-inspired” materials.2
References:
1. T. Coradin,
P. J. Lopez, ChemBioChem, 2003, 4,
251-259.
2. T. Coradin,
N. Nassif, J. Livage, Appl. Microbiol.
Biotechnol., 2003, 61, 429-434.
Protein-coated microcrystals: Applications in
biocatalysis
Michaela Kreiner, Norah O’ Farrell, Marie Claire
Parker
Dept. of Chemistry, University of Glasgow, Glasgow, G12 8QQ,
UK. E-mail: mickr@chem.gla.ac.uk
Enzymes frequently exhibit poor
activity in organic media when compared to their respective activities in
aqueous solution.[1] To overcome this problem, many strategies aimed
at optimising enzyme activity, such as manipulation of the micro-environment
and a range of immobilisation strategies have been studied. Whilst studying how
different methods of protein dehydration affected enzyme activity a new mode of
self-assembly was discovered. This new mode of self-assembly leads to the
formation of well-organised protein layers on the surface of micron-sized
crystals of water-soluble excipients e.g.
salts, sugars or amino acids. The crystals are produced in a one-step process:
a saturated aqueous solution of the excipient and protein is added drop-wise to
a suitable water miscible organic solvent. Instantaneous co-precipitation occurs
and the crystal lattice energy forces the protein molecules to migrate to the
surface of the excipient crystals.[2] We termed this novel system:
protein coated micro-crystals (PCMC).
|
Figure 1. Scanning Electron Microscopy image of
lipase coated micro-crystals.
|
Figure 2. 3D Topographical AFM image of a single protein coated
micro-crystal. Scan size: 400nm x 400nm
|
To date, we have studied this system
using (i) proteases and (ii) lipases. Catalytic activity measurements showed
that (a) the protein-coated crystals exhibited significantly higher activity
than their freeze-dried counterparts when assayed under the same conditions in
the organic solvent and (b) the crystals re-dissolved instantly in aqueous
buffer without loss of the original aqueous activity. This rapid dehydration
method leads to good retention of native structure (as shown by active site
titration) and Tapping Mode Atomic
Force Microscopy (AFM) images have confirmed that the protein layer is located
at the surface of the crystal and is easily accessible to the
titrant/substrate. Extending the choice of crystalline core material allows
fine-tuning of the biocatalyst system. For example, use of solid-state buffers
as crystalline matrix increases the activity of proteases and simultaneously
provides buffering capacity.[3]
This precipitation method leads to
the formation of a fine suspension (typically 0.1-5 mm for K2SO4
as carrier) with the protein-coated micro-crystals homogenously dispersed in
the organic solvent.[2] Such PCMC suspensions are easy to handle and
can be dispensed accurately as a suspension. This together with their high
storage stability at room temperature and high activity makes them favourable
candidates for biocatalyst screening programmes.
References
[1] A. M. Klibanov, Trends Biotechnol. 1997, 15, 97.
[2] M. Kreiner, B.D. Moore, M.C. Parker, Chem Commun, 2001, 1096.
[3] M. Kreiner, M.C. Parker, Biotechnol Bioeng, 2004,
accepted.
Labeling and immobilizing proteins via intein mediated ligation
Cameron Neylon, Robert Wood, Peter
Roach
School of
Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ,
Fax: +44 (0)23
8059 6805, e-mail: D.C.Neylon@soton.ac.uk.
Intein-mediated ligation
provides a site-specific method for the attachment of molecular probes to proteins.
The method is inherently flexible with regard to either the protein sequence or
the attached probe or solid support, but practical difficulties have limited
the widespread use of this valuable labeling system for the attachment of
small- to medium-sized molecules. We report studies to improve the efficiency and
practical application of these reactions, including the assembly of plasmids
for the expression of target-intein
fusion proteins and the analysis of their reaction with a fluorescent cysteine
derivative under a range of conditions. Optimal ligation of the fluorophore to
the target protein is critically dependent on the degree of oxidation of the fluorescent
cysteine derivative. Efficient ligation has been achieved with freshly prepared
fluorescent cysteine derivative under rigorously anaerobic conditions. Similar
ligation yields have also been achieved using more practically convenient
conditions including anaerobic reaction with addition of thiophenol, or aerobic
reaction with the further addition of tricarboxyethylphosphine.
References:
1. Muir TW, Sondhi D, Cole PA, Proc. Natl. Acad. Sci. U.S.A 1998,
95, 6705-6710
2. Wood RJ, Pascoe DD, Brown ZK, Medlicott EM,
Kriek M, Neylon C, Roach PL, Bioconjugate
Chemistry 2004, in press
De Novo Design as a Tool to Investigate
Protein Chemistry
Cecilia Tommos
Department of Biochemistry and Biophysics, Arrhenius
Laboratories for Natural Sciences, Stockholm University, SE-106 91 Stockholm,
Sweden; e-mail: cecilia@dbb.su.se
The biological functions of proteins are multifaceted
and range from providing cellular building material to catalyzing chemical
reactions to acting as gatekeepers to control the flux of ions and other molecules
across the cell membrane. Today we are limited in our knowledge of connecting
structural information to the functional properties of a protein, thus
stimulating increased research in this area. Ideally, one day it will be
possible to look at the amino-acid sequence of a protein and from this
information alone predict its three-dimensional structure and deduce its
function.
Varies approaches are being taken to
address the protein “structure-to-function” problem. The most common method is
to investigate structural and functional relationships of natural proteins by,
for example, site-directed mutagenesis studies. An alternative approach has
emerged in recent years involving the construction of proteins from scratch, so
called de novo protein design. The
goal with the de novo protein
approach is to derive design rules on how to create new, man-made proteins with
specific catalytic and biological functions.
We have developed two a-helical model proteins denoted a3W and a4W. a3W has
been characterized in some detail while the development of a4W is
more recent. To illustrate the versatility of de novo design in protein research, three different studies of the a3W
system will be described. In the first study we use a combination of
experimental and theoretical methods to investigate the redox properties of a3W. The second project involves using a3W to
examine the interaction energy of π-cation pairs. π -cation
interactions between aromatic (Phe, Tyr, Trp) and basic (His, Lys, Arg)
residues are common in natural proteins, although the strength of these
interactions has thus far been poorly characterized experimentally. We have
used high pressure to determine the energy of a Trp/Lys π -cation
interaction in a3W. With this technique we
avoid the uncertainties associated with introducing mutations. Finally, a
method will be described in which protein folding is driven by increasing the
energy of the unfolded state rather than lowering the energy of the folded
state.
Directed evolution on transketolase for obtaining new monosaccharides
V. Hélaine, A. Sevestre, A. Lasikova,
L. Hecquet.
Laboratoire SEESIB, UMR
6504 du CNRS, Ensemble scientifique des Cézeaux, 24 avenue des Landais, 63177
Aubière, France, Fax: +33 (0)473407717,
e-mail:
helaine@chimie.univ-bpclermont.fr.
During several years, we used
capabilities (enantioselectivity, stereospecificity) of transketolase (TK) for
obtaining many D-threo ketoses of biological interest (scheme 1a).1 Nevertheless, like many other enzymes, substrate specificity is
limited. We decided to evolve this enzyme in order to obtain D-threo
aldoses or L-erythro ketoses (non natural configuration) (Scheme 1b).
Thus, we used molecular biology techniques, especially random mutagenesis, on
wild type TK gene. After expression of the protein, we obtained a library of
different modified TK that have to be screened to select the most powerful
enzyme we are looking for.
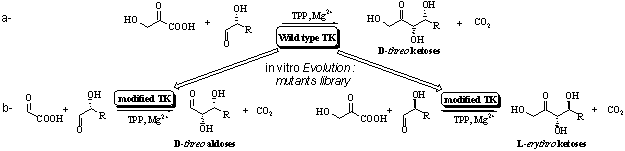
Scheme 1
We finalized a
qualitative and quantitative screening test on wild type TK using as a
substrate a ketose of natural configuration (D-threo) bearing
umbelliferone (scheme 2).2 Wild type TK is
able to recognize this substrate, then to clive it thus releasing
umbelliferone, a highly fluorescent compound.
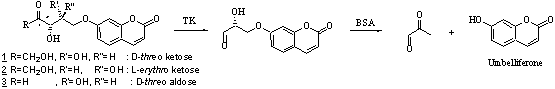
Scheme 2
We are going to
use this screening test for searching modified TK able to catalyze synthesis of
D-threo aldoses or L-erythro ketoses. For that purpose we are
preparing the corresponding substrates by chemically and/or enzymatically
routes.
References:
1. a) Guérard, C.; Alphand, V.;
Archelas, A.; Demuynck, C.; Hecquet, L.; Furstoss, R.; Bolte, J.; Eur. J.
Org. Chem. 1999, 1, 3399-3402. b) Crestia, D.; Guérard, C.;
Veschambre, H.; Hecquet, L. ; Demuynck, C.; Bolte, J.; Tetrahedron:
Asymmetry 2001, 12, 869-876
2. Sevestre, A.; Hélaine, V.;
Guyot, G.; Martin, C.; Hecquet, L.; Tetrahedron Lett. 2003, 44,
827-830
Taming Giants: Insights into DNA Polymerase Function
Andreas Marx
Universität
Konstanz, Fachbereich Chemie, Universitätsstrasse 10, 78457 Konstanz, Germany,
Fax: +49 (0)7531 885140, e-mail: Andreas.Marx@uni-konstanz.de.
One essential prerequisite of any organism is to keep its
genome intact and to accurately duplicate it before cell division. All cellular
DNA synthesis required for DNA repair, recombination, and replication depends
on the ability of DNA polymerases to recognize the template and insert the
canonical nucleotide. A DNA polymerases is presented with a pool of four
structurally similar deoxynucleotide triphosphates (dNTPs) from which it must
select the sole correct (i.e., Watson-Crick base paired) substrate for
incorporation into the growing DNA strand. This leads to the question: Which
properties could enable a DNA polymerase to catalyze nucleotide incorporation
with a selectivity far greater than that which is dictated by the thermodynamic
differences between base pairs in free solution? Using chemically engineered
tools, we were able to gain new insight into these complex enzymatic processes.[1]
Apart from their pivotal role
in biological process DNA polymerases are widely applied in numerous molecular
biological and biotechnological applications. After the completion of the human
genome sequence the discovery of small dissimilarities in the sequence of
different individuals is one of the prime tasks today, since genetic
differences may influence the variability of patients’ response to drugs.
Highly selective DNA replication systems should simplify the detection of
single nucleotide polymorphisms (SNP) in genes without need for further tedious
time- and costs-consuming analytical procedures as applied to date. In the talk
chemical and genetic approaches towards achieving DNA polymerase based systems
with improved selectivity for efficient genome-typing will be discussed.[2]
References:
1. a) D.
Summerer, A. Marx, Angew. Chem. 2001, 113,
3806-8; b) D. Summerer, A. Marx, J. Am. Chem. Soc. 2002, 124,
910-1; c) M. Strerath, D. Summerer, A. Marx, ChemBioChem 2002, 3,
578-580. d) M. Strerath, J. Cramer, T. Restle, A. Marx, J. Am. Chem. Soc. 2002, 124, 11230-11231.
2. a) M. Strerath, A. Marx, Angew. Chem. Int. Ed. 2002, 41, 4766-69;
b) M. Strerath, J. Gaster, D. Summerer, A. Marx ChemBioChem 2004, 5, 333-9; c) A.
Marx, D. Summerer, N.Z. Rudinger, patent application.
Synthesis of Hydroxamic Acid in TTAB Reversed Micelles using Amidase
from Pseudomonas sp.
R. Pachecoa,b, A. Karmalib, M.L.
Serralheiroa
a Faculdade de Ciências da
Universidade de Lisboa-Departamento de Química e Bioquímica, Campo Grande
1749-016 Lisboa, Portugal e-mail: rpacheco@deq.isel.ipl.pt
b Instituto Superior de Engenharia de
Lisboa, R. Conselheiro Emídio Navarro. 1949-014 Lisboa, Portugal
Hydroxamic acids
derivatives have a wide spectrum of application due to their ion metal
chelating capacity, mainly their capacity to inhibit metalloproteinases that
are involved in human diseases [Fournand, 1997]. These compounds are used in
several pharmaceutical drugs. Amidases (E.C. 3.5.1.4) are enzymes that in
nature catalyse the hydrolysis of amide bonds in small aliphatic amides but
these enzymes also have the capacity to catalyse the acyl group transferase to
amines what enlarged the possibility of synthesising several hydroxamic acids
derivatives if the acyl acceptor is hydroxylamine. Amidases with this capacity
have been isolated mainly from Pseudomonas
and Rhodococcus strains [Brown, 1973
and Fournand 1998]. As the amidase catalysed reaction involves a ping-pong
bi-bi mechanism [Fournand, 1998] with the formation of acyl-enzyme complex that
can transfer the acyl unit either to an amine or to the water present in the
system, it is the kind of reaction to be performed in organic medium in order
to diminish the presence of water. Reverse micelles formed in organic medium
have been used to encapsulate enzymes in order to catalyse the synthesis of
amide bonds with the formation of dipeptides [Serralheiro 1994]. In these
systems the enzyme remains soluble, what avoids problems with mass transfer
that could slow down the reaction velocity and they can easily be transformed
into a continuous reaction system. In this work the amidase from a Pseudomonas aeruginosa gene expressed in
E. coli, was purified and
encapsulated in reverse micelles of the cationic surfactant
tetradecylltrimethyl ammonium bromide dissolved in heptane/octanol. The effect
of several parameters of the system that could influence the enzymatic activity
were analysed. It is the results of this study that are to be presented.
References:
1. D. Fournand, F. Bigey, F. Ratomahenina, A.
Arnaud and P. Galzy (1997) Biocatalyst improvement for the production of
short-chain hydroxamic acids Enzyme
Microbial Techology, 20,
424-431.
2. P.R. Brown, M.J. Smyth, P.H. Clarke and M.A.
Rosemeyer (1973) The subunit structure of the aliphatic amidase from Pseudomonas aeruginosa. Eur J. Biochem. 34, 177-187.
3. D. Fournand, F. Bigey and A. Arnaud (1998)
Acyl transfer activity of an amidase from Rhodococcus
sp. Strain R312: Formation of a wide range of hydroxamic acids. Applied and Environmental Microbiology 64, 2844-2852.
4. D. Fournand, A. Arnaud and P. Galzy (1998)
Study of the acyl transfer activity of a recombinant amidase overproduced in a E.coli Strain. Application for
short-chain hydroxamic acid and acid hydrazide synthesis. Journal of Molecular Catalysis B: Enzymatic 4, 77-90.
5. M.L.M. Serralheiro and J.M.S. Cabral (1994) Synthesis of AcPheLeuNH2 by chymotrypsin in TTAB reversed
micelles…optimization of the system. Biotechnol.
Bioeng. 43, 1031.
Modelling the active site of Nitrile Hydratase: oxidation of (N2S2)-Fe
and (N2S2)-Co complexes
Erwan Galardon, Sandrine
Chatel, Mathieu Rat, Isabelle Artaud*
UMR 8601, Université René Descartes, 45 rue des Saints
Pères, 75270 Paris Cedex 06, France; e-mail: erwan.galardon@univ-paris5.fr
Nitrile Hydratases (NHases) are bacterial
metalloenzymes which catalyze the hydratation of nitriles to the corresponding
amides. Their active site contains a low spin non-heme iron(III) or non-corrin
cobalt(III) center. Two carboxamido nitrogens, one cysteine-sulfur, and two
oxidized sulfurs (one cysteine-sulfinic and one cysteine-sulfenic acid)
constitute the donor set around the metal, in addition to a water/hydroxide
molecule in the active form. This unique coordination sphere, which has
implications on the mecanism of the hydratation, has pushed several groups1
to design synthetic models of the catalytic center of iron and cobalt NHases.
Results obtained in our group in recent years will be discussed.
References:
1. Harrop,
T. C. and Mascharak, P. K., Acc. Chem. Res. ACS ASAP; Kovacs, J. A., Chem. Rev. 2004, 104, 825-848.
Designing of Mn complexes as SOD
synzymes. Evaluation of the SOD-activity: from anhydrous experiments to
transient kinetics in aqueous solution.
Clotilde
Policara, Stéphanie Durota, François Lamberta,
Jean-Pierre Mahya, Jean-Philippe Renaultb, Giorgio Pelosic
a Laboratoire de Chimie Biorganique et
Bioinorganique, CNRS-UMR8124, Université Paris XI, F-91405 ORSAY cedex, France
cpolicar@icmo.u-psud.fr
b Laboratoire
de Radiolyse, CEA-Saclay, , F-91191 GIF-sur-YVETTE cedex, France
c
Dipartimento di Chimica Generale ed Inorganica, Chimica Analitica, Chimica
Fisica, Parco Area delle Scienze 17A, Universita'
di Parma, 43100 Parma, Italy.
Reactivity of superoxide is of current interest as it plays a key role
in oxidative stress. Superoxide dismutases (SODs) are metalloenzymes involved
in the protection of the cell against superoxide. Elucidation of reaction
mechanisms of superoxide with metal centers, both at the active site of
superoxide dismutases and with inorganic complexes is important to design efficient
synthetic compounds valuable as pharmaceuticals. A family of Mn(II) and Mn(III)
complexes have been synthesised based on N-tripodal ligands (see figure). Their
structures have been solved, showing a large variety of structures in the solid
state: inorganic polymers with bridging carboxylatos to dimer and monomer.(1,2,3)
The reactivity towards superoxide has been studied, both in anhydrous medium
and in aqueous buffers.(2) In anhydrous medium, EPR, cyclic
voltammetry, low-temperature UV-visible spectroscopy were used to seek for
intermediates species or species directly derived. Dimeric species where shown
to be produced. In aqueous buffer, the Fridovitch-McCord test showed that this
series of complexes display SOD-activity, by comparison to the literature (kcat
from 3 106 to
7 106 M-1s-1). Reactivity of two of these
complexes has been studied by pulsed radiolysis. It provided kinetics constant
in agreement with that from Fridovith-McCord test. Moreover, unambiguous
evidences for catalytic activity were obtained. A mechanism has been proposed
that is consistent with all experimental data.
This study shows the complementarity of experiments in anhydrous and
aqueous buffer.
References:
(1) Policar, C.; Lambert, F.; Cesario, M.;
Morgenstern-Badarau; I. Eur. J. Inorg.
Chem., 1999, 2201-2208.
(2) Policar,
C.; Durot, S.; Lambert, F.; Cesario, M.; Morgenstern-Badarau; I. Eur. J. Inorg. Chem., 2001, 1807-1818.
(3) Durot, S.; Policar, C.; Pelosi, G.;
Bisceglie, F.; Mallah, T.; Mahy, J.-P. Inorg.
Chem., 2003, 42, 8072-8080.
From Enzyme Models to Synthetic Proteins: a Chemist’s Approach to the
Study of Enzymes
Gerard Roelfes
University
of Groningen, Departments of Organic Chemistry and Biochemistry, Nijenborgh 4,
9747 AG Groningen, the Netherlands, email: Roelfes@chem.rug.nl
In this talk I
wish to demonstrate what a chemist can contribute to the understanding of
enzymes, in particular by using the power of synthesis.
The first part
will deal with the study of synthetic active site models. Enzyme models have
the advantage that they are readily available and easier to study than an
actual enzyme. The information obtained from these model compounds can help
increase our understanding of how enzymes work. Also these models can serve as
a starting point for the development of a new generation catalysts with
increased activity and selectivity. Our research has been focussed on iron
complexes with multi-dentate nitrogen ligands as models for non-heme iron
containing oxygenases. In the course of this study several iron-peroxide
intermediates have been characterized and their relation to the observed
catalytic activity has been established.
A main criticism
of enzyme models is that they’re models. This means that you can ask the
question: how relevant is the information you obtain from a model compounds
when you disregard the protein matrix in which the active site is embedded? To
address this we make proteins containing non-natural amino acids as mechanistic
or spectroscopic probe, so we can get information from the actual protein.
Selenium is ideal as a mechanistic probe since it is isosteric to sulphur. So
it can be introduced in the form of selenocysteine or selenomethionine and thus
cause minimal structural perturbation. In the second part of the talk I will
discuss (semi-) synthetic strategies to selenium containing proteins and the
application of selenium as a mechanistic and/or NMR probe.
Modulation of Chirality of Lipases via immobilization techniques
Jose M. Palomoa,b, R. Fernández-Lafuentea,Jose M. Guisána
a Departamento de
Biocatalisis. Instituto de Catalisis (CSIC) Campus UAM Cantoblanco, 28049 Madrid, Spain, e-mail: josempalomo@icp.csic.es,
rfl@icp.cisc.es, josemguisan@icp.csic.es
b current address: Deparment of Chemical Biology.Max Planck Institute of Molecular Physiology Otto-Hahn st 11 Dortmund (Germany). jose.palomo@mpi-dortmund.mpg.de
References:
Palomo,
Jose M.; Fernández-Lorente, G.;
Mateo, C.; Fuentes, M.; Fernández-Lafuente, R.; Guisán, JM. Tetrahedron: Asymmetry 2002, 13, 1337-1345.
Palomo, Jose M.; Fernandez-Lorente, G.; Mateo,C.; Ortiz,C.;
Fernandez-Lafuente,R.; Guisan J,M.. Enzyme Microb Technol. 2002,
31, 775-783.
Palomo Jose M.; Muñoz, G.; Fernández-Lorente, G.; Mateo,
C.; Fuentes, M.; Guisán J.M.; Fernández-Lafuente, R. J. Mol. Cat B: Enzymatic 2003,
21, 201-210.
Palomo, Jose M.; Fernandez-Lorente,
G.; Rúa, M.L.; Guisan,J.M.; Fernández-Lafuente,R. Tetrahedron: Asymmetry 2003, 14, 3679-3687.
Palomo, Jose M.; Fernández-Lorente,G.;
Mateo,C.; Fernández-Lafuente ,R.; Guisan, J. M. Tetrahedron: Asymmetry 2002,
13, 2375-2381.
Palomo, Jose M.; Mateo,C.;
Fernández-Lorente,G.; Solares , L.F.; Diaz, M.;. Sánchez, V.M.;
Bayod,M.; Gotor, V.; Fernandez-Lafuente
,R.;Guisan , J.M. Tetrahedron: Asymmetry 2003, 14, 429-438.
Palomo Jose M.; Segura, R.L; Fernández-Lorente, G.;
Guisán J.M.; Fernández-Lafuente,R. Tetrahedron:
Asymmetry 2004; in press
Where
Bioinorganic Chemistry Meets Molecular Architecture: Modelling Metal-Nucleic
Acids Interactions
Bernhard Lippert
Fachbereich Chemie, Universität
Dortmund, Otto-Hahn-Str. 6, 44221 Dortmund, Germany¸ Fax: +49 (0)231 755
3797; e-mail: bernhard.lippert@uni-dortmund.de.
Metal ions are
natural counter ions of the polyanionic nucleic acids. Depending on the nature
of the metal and its environment by nucleobase donor atoms, metal-nucleic acid
interactions can range from non-specific to highly specific ones. Although
basic metal binding patterns with nucleic acid constituents are reasonably well
understood,1 the role of metal ions in special tertiary
interactions, especially with folded RNA structures and multistranded DNAs are
still a topic of great interest. Applying non-physiological metal ions,
artificial analogues of natural base-metal aggregates can be synthesized and
tested for potential uses as chemical probes or novel chemotherapeutic agents2,3
other than the antitumor agent Cisplatin.4 Renewed interest in
metal-nucleobase interactions stems from the suspected or proven role of metal
ions in RNA catalysis.5 Apart from a direct involvement of
metals in such reactions, there is also the possibility of an indirect
role of metal ions by initating acid-base catalysis as a consequence of
profound perturbations of the normal pKa
values of nucleobases following metal coordination.6
References:
1. B. Lippert, Coord. Chem. Rev., 2000,
200-202, 487.
2. B. Lippert and M. Leng, Topics
in Biol. Inorg. Chem.,
Vol. 1: Metallapharmaceuticals, M. J. Clarke and P. J. Sadler (Eds),
Springer, Heidelberg, 1999, pp. 117 – 142.
3. E. Freisinger, I. B. Rother, M. S. Lüth, and
B. Lippert, Proc. Natl. Acad. Sci. USA, 2003, 100, 3748.
4. B. Lippert (Ed.), Cisplatin-Chemistry
and Biochemistry of a Leading Anticancer Drug, VHCA Zürich and Wiley-VCH,
Weinheim 1999.
5. A. M. Pyle, J. Biol. Inorg. Chem., 2002,
7, 679 and refs. cited.
6. (a) M. Garijo Añorbe, M. S.
Lüth, M. Roitzsch, M. Morell Cerdà, P. Lax, G. Kampf, H. Sigel, and B. Lippert,
Chem. Eur. J., 2004, 10, 1046. (b) M. Roitzsch and B.
Lippert, J. Am. Chem. Soc., 2004, 126, 2421.
 CERC3 Young Chemists'
Workshop "Biocatalysis"
CERC3 Young Chemists'
Workshop "Biocatalysis"
March 24 - March 27,
2004
Institut für Organische
Chemie, Henkestr. 42, 91054 Erlangen, Germany

Poster
Titles
(only main authors)
P1 Mahdi Abu-Omar
“Investigations
of nitric oxide reactions with non-heme phenylalanine”
P2 Frank Bordusa
“Old Enzymes with new synthetic properties”
P3 Thibaud Coradin
“Bacteria
encapsulation in sol-gel silica matrices”
P4 Paul Dalby
“Expanding the synthetic repertoire of transketolase”
P5 Erwan Galardon
“Modelling the active site of Nitrile Hydratase:
oxidation of (N2S2)-
Fe and (N2S2)-Co
complexes”
P6 Brian Gibney
“Heme Protein Biocatalysis: Lessons
from the Protein Data Bank and DeNovo
Heme Protein Design”
P7 Craig Grapperhaus
“Synthesis
and Characterization of N2S3X-Fe Models of Iron-Containing Nitrile
Hydratase”
P8 Virgil
Hélaine
“Directed
evolution on transketolase for obtaining new monosaccharides”
P9 Michaela Kreiner
“Protein-coated
micro-crystals”
P10 Andreas Marx
“Taming giants: Insights into
DNA Polymerase Function”
P11 Cameron Neylon
“Labeling
and immobilizing proteins via intein mediated ligation”
P12 Sandrine
Ollagnier de Choudens
“Recent developments on biotin synthase”
P13 Rita Pacheco
“Biocatalysis
of hydroxamic acid using reverse micelles”
P14 Jose M. Palomo
“Modulation
of lipases chirality via different immobilization techniques”
P15 Tatyana
Polenova
“Solid-State
NMR Spectroscopy for Studies of Geometry and Electrostatic
Environment
in Vanadium Haloperoxidases”
P16 Clotilde Policar
“Imidazole and
imidazolate iron complexes: on the way for tuning 3D-structural characteristics
and reactivity. Redox interconversions controled by protonation state”
P17 Markus Reiher
“DFT calculations of spin
states and spin barriers in transition metal catalysis”
P18 Gerard Roelfes
“(Semi-)Synthetic
Enzymes”
P19 Grit Straganz
“Structure-activity
relationships of the non-heme iron dependent dioxygenase Dke1”
P20 Roderich Süssmuth
“Abyssomicins – novel inhibitors of the p-aminobenzoic
acid/tetrahydrofolate
biosynthesis”
P21 Cecilia Tommos
“De
Novo Design as a Tool to Investigate Protein Chemistry”
P22 Andrea Zocchi
“Artificial
metalloenzymes for enantioselective catalysis based on biotin-avidin
technology”
 CERC3 Young Chemists'
Workshop "Biocatalysis"
CERC3 Young Chemists'
Workshop "Biocatalysis"
March 24 - March 27,
2004
Institut für Organische
Chemie, Henkestr. 42, 91054 Erlangen, Germany

Authors
|
Author
|
Page
|
Author
|
Page
|
|
Abu-Omar,
Mahdi M.
|
9
|
Martinez,
Alexandre
|
13
|
|
Amoroso,
Jennifer H.
|
20
|
Marx,
Andreas
|
30
|
|
Arends,
Isabel
|
12
|
Mashuta, Mark
S.
|
16
|
|
Artaud, Isabelle
|
32
|
Meunier,
Bernard
|
13
|
|
Asano,
Yasuhisa
|
21
|
Müller,
Michael
|
19
|
|
Bischoff,
Daniel
|
11
|
Mulliez, E.
|
22
|
|
Bister, Bojan
|
11
|
Neylon,
Cameron
|
27
|
|
Bordusa,
Frank
|
21
|
Nidetzky,
Bernd
|
17
|
|
Bull, Alan
T.
|
11
|
O’ Farrell,
Norah
|
26
|
|
Chatel, Sandrine
|
32
|
Ollagnier-de
Choudens, Sandrine
|
22
|
|
Collot, Jérôme
|
24
|
Pacheco, Rita
|
31
|
|
Coradin, Thibaud
|
25
|
Palomo, Jose
M.
|
35
|
|
Dalby, Paul
A.
|
18
|
Parker, Marie
Claire
|
26
|
|
Davis,
Benjamin G.
|
14
|
Patra, Apurba
K.
|
16
|
|
Durot, Stéphanie
|
33
|
Pech,
Andreas
|
21
|
|
Ebel, Martin
|
10
|
Pelosi, Giorgio
|
33
|
|
Fernández-Lafuente,
R.
|
35
|
Pöhlmann,
Angela
|
21
|
|
Fiedler,
Hans-Peter
|
11
|
Pooransingh,
Neela
|
10
|
|
Fontecave, M.
|
22
|
Polenova, Tatyana
|
10
|
|
Francesconi,
Lynn C.
|
10
|
Policar, Clotilde
|
33
|
|
Galardon, Erwan
|
32
|
Poturovic,
Selma
|
16
|
|
Gibney,
Brian
|
20
|
Rall, Kathrin
|
21
|
|
Goodfellow,
Michael
|
11
|
Rat, Mathieu
|
32
|
|
Gradinaru, Julieta
|
24
|
Reedy,
Charles J.
|
20
|
|
Grapperhaus,
Craig A.
|
16
|
Rehder, Dieter
|
10
|
|
Grogan,
Gideon
|
23
|
Reicke,
Andreas
|
11
|
|
Guisán, Jose
M.
|
35
|
Reiher,
Markus
|
15
|
|
Hélaine, Virgil
|
29
|
Renault, Jean-Philippe
|
33
|
|
Hemmert,
Catherine
|
13
|
Riedlinger,
Julia,
|
11
|
|
Hecquet, L.
|
29
|
Roach,
Peter
|
27
|
|
Huang, Wenlin
|
10
|
Roelfes,
Gerard
|
34
|
|
Humbert, Nicolas
|
24
|
Rubach, J.
|
22
|
|
Jantzen, Sven
|
10
|
Schmidt,
Stephanie
|
21
|
|
Karmali, A.
|
31
|
Serralheiro,
M. L.
|
31
|
|
Klein, Gérard
|
24
|
Sevestre, A.
|
29
|
|
Klußmann,
Sven
|
21
|
Sheldon,
Roger
|
12
|
|
Komeda,
Hidenobu
|
21
|
Skander, Myriem
|
24
|
|
Kozlowski,
Pawel M.
|
16
|
Süssmuth,
Roderich D.
|
11
|
|
Kreiner, Michaela
|
26
|
Straganz,
Grit
|
17
|
|
Lasikova, A.
|
29
|
Tommos, Cecilia
|
28
|
|
Lambert, François
|
33
|
Ward, Alan
C.
|
11
|
|
Leonard,
Philip M.
|
23
|
Ward, Thomas R.
|
24
|
|
Li, Ming
|
16
|
Wehofsky,
Nicole
|
21
|
|
Li, Yu-Xin
|
12
|
Whittingham,
Jean L.
|
23
|
|
Lippert,
Bernhard
|
36
|
Wood, Robert
|
27
|
|
Livage, Jacques
|
25
|
Zähner,
Hans
|
11
|
|
Loosli, Andreas
|
24
|
Zgierski,
Marek Z.
|
16
|
|
Mahy, Jean-Pierre
|
33
|
Zhuang,
Jinyou
|
20
|
|
Maldonado,
Louis A.,
|
11
|
Zocchi, Andrea
|
24
|