A comparison of theory and experiment.
by Olga Dmitrenko, a) * Robert D. Bach,a) Rafal R. Sicinskib) and Wolfgang Reischlc)
a) Department of Chemistry and Biochemistry, University of Delaware, DE, USA
b) Department of Chemistry, University of Warsaw, Warsaw, Poland
c) Department of Chemistry, University of Vienna, Vienna, Austria
Introduction
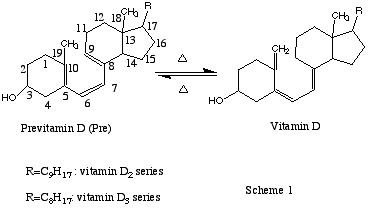
One of the key and final reaction of the in vivo and in vitro vitamin D synthesis is thermal interconversion previtamin D <-> vitamin D (Scheme 1) via the [1,7]-antarafacial hydrogen shift.1a The effects of fluorine and methoxy or acetoxy substituents at C-19 on the [1,7]-hydrogen migration in previtamin D analogs have been studied experimentally in the last twenty years by several groups in the USA and in Europe. In all these studies, it has been found that a 19,19-difluoro substitution completely inhibits the reaction, whereas a 19-methoxy (-acetoxy) substituent accelerates the conversion of previtamin D to vitamin D.1,2
In spite of the experimental agreement, the mechanistic interpretations of these opposite effects were controversial.1,2 Sialom and Mazur1c suggested two alternative explanations for the reaction accelerating effect of the 19-acetoxy substituent. One involves the electron donating property of oxygen atom, which changes the electron distribution in the triene system, and the second is a steric effect that may destabilize the s-cis 6,7 conformation of the previtamin D molecule. The complete inhibition of thermal isomerization for 19,19-difluoro substitution was explained by strong inductive effect of the two fluorine atoms1c based on analogy to [1,5]-H shift reactions3 and the assumption that a localized positive charge on the terminal carbon atom of the triene system is involved in the transition structure for [1,7]-hydrogen migration.
However, in other studies,2b it has been concluded that inductive effects do not play an important role in the stability of 19-acetoxy and 19-methoxy vitamin D analogs. It was also questioned whether the thermal stability of 19,19-difluoroprevitamin D can be attributed to the strong inductive effect2b of the fluorine atoms.
More recently, effects of electron-donating methoxy substituents and electron-withdrawing fluoride substituents on the [1,5]-H shift were studied in several model 1,3-pentadienes.4a It was suggested that decreasing the electron density of the p-system destabilizes the aromatic TS and increases the activation energy. Importantly, it was suggested that not only inductive effects, but also orbital-orbital and steric interactions may affect the energetics of this pericyclic reaction.4a
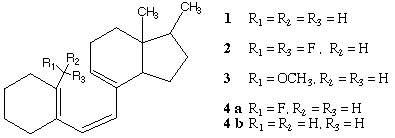
In order to clarify the mechanisms controlling the reaction barriers for the [1,7]-H shift in previtamin D model 1 and its C-19 modified analogs 2-4 (substituted at C-10 with CH2F, CHF2 and CH2OCH3 group, see Scheme 1), we performed density functional studies using the B3LYP hybrid method5. This method produces satisfactory results4b for the experimental activation energies for the [1,5]-H shift in 1,3-pentadiene and antrafacial [1,7]-H shift in previtamin D (EA=19.5 kcal/mol4c) systems as discussed in ref.4b.
 Scheme
1
Scheme
1
The growing awareness of the importance of the interactions between filled and unfilled orbitals6 has encouraged us to examine the possible correlation between relative energies of natural bond orbitals (NBO), orbital interactions7 and the activation barriers for the C-19 substituted systems.
Computational Methods
Density functional studies and ab initio molecular orbital calculations8 were performed with Gaussian 98 system of programs.9 For the NBO analysis, the NBO 4.0 module of Gaussian 98 was used. The Becke three-parameter hybrid functional5a,c combined with the Lee, Yang and Parr (LYP) correlation functional,5b denoted B3LYP,5d was employed in the calculations using density functional theory (DFT). Geometries were optimized10 at the B3LYP level using the 6-31G(d) basis set. In the smallest model, we applied also B3LYP/6-31+G(d,p) and CAS(6,6)/6-31G(d)+MP2 methods. For the non-substituted system, all stationary points were characterized by frequency calculations. Homolytic bond energies (DE) quoted in the text are derived from G2 total energies while bond dissociation energies (BDE) are derived from DH(298).11
Results and discussion
Three lowest energy transition structures (TS-1, TS-2, TS-3, Figure 1) and the corresponding previtamin D model minima12 (GS-1, GS-2, GS-3) have been fully optimized.13 Their selected geometrical characteristics are given in Table 1.
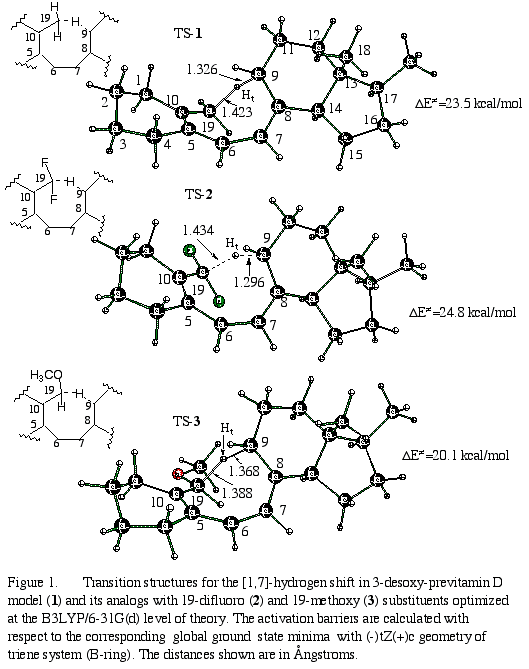
Table 1. Selected
geometrical data on B3LYP/6-31G* optimized ground-state minima (GS) and
transition structures (TS) for [1,7]-H shift in 1, 2 and
3
model systems.
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a) in the
global minimum with (-)tZ(+)c geometry. b)
in the precursors
for the TS in the folded conformations (+)cZ(+)c.
The comparison of the
distances between migrating hydrogen (Ht) and carbon atoms 9
and 19 in the transition structures suggests that TS-3 is an early
TS, whereas TS-2 is a late transition state. This suggestion is
supported by the relative calculated activation barriers (given in Figure
1) for the exothermic [1,7]-H shifts.14
The reaction in the
parent model system 1 (R1=R2=H) has a classical
barrier of 23.5 kcal/mol (Table 2). This value is about 4 kcal/mol higher
than experimental activation barrier. The activation entropy is twice smaller
than experimental (-20.4 e.u.)4c. The increase in basis set
does not affect the activation barrier and activation entropy as it is
seen from the B3LYP/6-31+G(d,p) results for the smallest model (second
row, Table 2). CAS(6,6)+MP2 results suggest that B3LYP underestimates the
barrier by about 3 kcal/mol. Nevertheless, for an insight on the possible
electronic/orbital effects, we think that B3LYP/6-31G(d) method is an adequate
selection for the treatment of such large molecules (the presence of the
5-membered D-ring in the models is necessary since it affects the energy
gap between right- and left-handed TSes13).
Relatively to parent
system, 19-difluoro substitution increases the reaction barrier to DE/
=24.8 kcal/mol, whereas a 19-methoxy reduces the barrier by 4.7 kcal/mol
(DE/=20.1
kcal/mol). These changes of the reaction barrier caused by substituents
are in full agreement with the experimental observations1 that
methoxy-substituent significantly accelerates the reaction whereas difluoro
substitution completely inhibits the hydrogen migration1c.
Table 2. Calculated
Activation Parameters for the [1,7]-H Shift in previtamin D models (B3LYP/6-31G(d),T=298.15°
K, scaling factor f=0.9989).
|
|
|
e.u. |
|
|
|
|
|
|
(20.87) |
(-9.25) |
(20.00) |
|
 |
20.52c |
15.31b |
-8.27 b |
14.30 b |
|
Surprisingly, the calculated
charges do not support these expectations. Both cases showed the same character
of changes in the charge distribution (Table 3). Moreover, in 19-difluoro
model 2, the net NBO charge on C-19 is positive for ground-state
minimum (0.6 e) and transition state (0.7 e), whereas in model
3,
these charges are still negative (as in model 1), but significantly
decreased (-0.1 e in GS-3 and -0.01 e in TS-3).
Table 3. NBO
charges (B3LYP/6-31G*) on carbon atoms of the triene system and migrating
hydrogen in the previtamin D model 1 and its 19-substituted analogs
(2 and 3) in the ground state (GS)a) and the corresponding
transition structures (TS).
|
10b -CH3 |
-CHF2 |
-CH2OCH3 |
10b -CH3 |
-CHF2 |
-CH2OCH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a) all the GS (ground state minimum) charge data are calculated for the (+)cZ(+)c conformers.
In the ground state minima GS-2, GS-3 and transition structures TS-2, TS-3, the charges on carbon atoms 5, 7 and 9 were increased relative to the parent model 1 (GS-1, TS-1), whereas on carbons 6, 8 and 10 they were decreased. Based upon the charges (Table 3), one may even expect that due to an electrostatic repulsion between leaving hydrogen (Ht) and positively charged C-19 (+0.6 in GS-2) the cleavage of C(19)æ H bond should be significantly facilitated in comparison with the two other models 1 and 3.
In the Natural Bond Orbital study of ground state (+c)Z(+)c conformers GS-1, GS-2 and GS-3, our interests were focused on C(8)-C(9) p-bond, C(19)-Ht s-bond and corresponding antibonding orbitals (antibonds) since they should be affected in the primary stage of the [1,7]-hydrogen shift. We found that in all three TSs there are two comparable stabilizing interactions between filled and unfilled orbitals of C(8)-C(9) and C(19)-Ht bonds, p-s* and s-p* interactions (Scheme 3). In both cases, difluoro- and methoxy-substitution, these energetic contributions are slightly larger than in parent system 1. Thus, this analysis does not provide an answer to the opposite effects in models 2 and 3, both substitution patterns contribute through filled/unfilled bond orbital interactions to the transition state stabilization similarly.
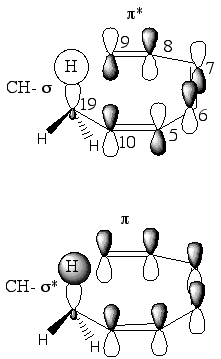 Scheme
3
Scheme
3
In Table 4, we summarized
the natural bond orbitals (NBO) energies for the C(8)-C(9) p-bond,
C(19)-Ht s-bond
and corresponding antibonding orbitals.
Table 4. Calculated
Natural Bond Orbitals and Energies (a.u.) for Selected Bonds in GS-1,
GS-2 and GS-3 (+)cZ(+)c-conformers, B3LYP/6-31G(d).
|
|
10b -CH3 |
-CHF2 |
-CH2OCH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
a) asterisk
indicates antibonding orbital; b) the energy difference between
interacting bonding and antibonding NBOs is given in kcal/mol,
DEps
* = E s*[C(19)-H]
- E p[C(8)-C(9)];
DEsp*
= Ep*[C(8)-C(9)]
- Es[C(19)-H].
The character of changes in GS-2 and GS-3 NBOs in comparison with GS-1 is quite similar, and does not provide an explanation for why difluoro- and methoxy-effects are opposite. Interestingly, the difluoro substiuent causes remarkable increase of the energy gap between filled C(19)-Ht s-orbital and unfilled C(8)-C(9) p-orbital (DDEsp*=37.0 kcal/mol) in contrast to the case of methoxy substitution (DDEsp*=4.3 kcal/mol).
This prompted us to
analyze the possible effect of both substituents on the C(19)H
bond energy at the G2 level of theory15 using small model molecules
(ethane, its difluoro and methoxy derivatives). The bond energy data given
in Table 5 suggest that the proximity of oxygen reduces the homolytic bond
dissociation energy (BDE) of CH by about 6 kcal/mol, whereas difluoro
substituent does not affect the CH bond energy.
Table 5. Calculated
Homolytic CH Bond Energies and Bond Dissociation Energies (kcal/mol)
at the G2(MP2) and G2 Levels of Theory for the Ethane Molecule and its
Difluoro- and Methoxy- Substituted Analogs.
|
|
|
|
|
D H° 298 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
This provides an explanation for the reduced barrier for the [1,7]-H shift in 19-methoxy substituted previtamin D (3) since the weaker C(19)H bond can be broken more easily (providing earlier TS) due to presence of neighboring oxygen. The comparison of the proton affinities (PA) of the H atom to be shifted (Ht) is also informative. The previtamin D model 1 in its (+)cZ(+)c conformation has estimated PA=389.5 kcal/mol at B3LYP/6-31G(d). At the same level, 19-methoxy model 3 has reduced PA=383.5 kcal/mol consistent with amore facilitated homolytic C(19)H bond cleavage. This is supported by NBO charge distribution as well. The reduction of the electron density around C19 in GS-3 and TS-3 in comparison with the parent system 1 is calculated (Table 2). Thus, more electronegative methoxy- substituent tend to destabilize the C(19)Hd+.4a
Based upon NBO analysis, that indicates a similar effect for both types of C(19) substitution (model 2 and 3), one may rather expect a decrease of the reaction barrier in both these cases. Inspection of the structure of TS-2 indicates that one of fluorine atoms is in close proximity to the triene system (the distances between F and olefinic carbon atoms are smaller than 2.9 Å). This may suggest that the repulsion between fluorine atom and negatively charged p-cloud over the triene system destabilizes the transition structure thereby rising the reaction barrier.16 In order to check this assumption, 19-fluoro monosubstituted model 4 has also been studied. We have optimized the global minimum GS-4 with (-)tZ(+)c conformation which is similar to those found for GS-1, GS-2 and GS-3, and in the same manner we have located TS-4a and TS-4b with in exo- and endo- positions of the fluorine atom with respect to the triene system (Figure 2).
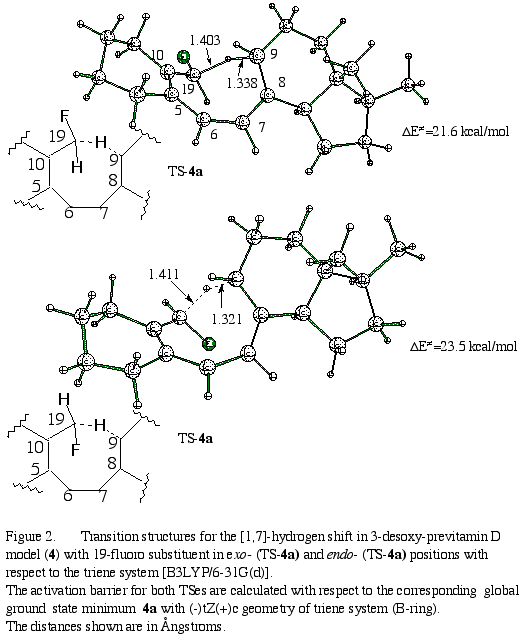
As expected from above
idea about the role of an electrostatic CF/p
repulsion, in the case of the model with one fluorine atom at C-19 in the
exo-position
(TS-4a), the reaction barrier is lowered with respect to difluoro-substituted
model
2
to 21.6 kcal/mol. The rotation of the 19-fluoro-substituend
toward the triene system into its endo-position destabilizes the
transition structure TS-4b (Figure 2) by about 2 kcal/mol. We believe
the presence of second fluorine atom in model 2 provides an additional
destabilization (about 1-2 kcal/mol) through long-distance electrostatic
interactions.
Conclusions.
Gas-phase B3LYP/6-31G(d)
calculations performed in this study suggest that 19-methoxy- and 19,19-difluoro-
substituent effects may have different origins (induced charge re-distribution
in the first case and electrostatic CF/p(triene
system) repulsion - in the former case). The comparisons of the G2 bond
dissociation energies for smaller model molecules, NBO charge distributions
in the model systems
1-3
and their corresponding transition
structures for the [1,7]-H shift do provide an explanation for the significant
acceleration of the reaction in 19-methoxy-previtamin, but fail to predict
the effect of inhibition by 19,19-difluoro- substitution. Calculated effect
of the transition structure destabilization in 19-fluoro-substituted previtamin
D model 4b with F-atom in endo-position vs model
4a
with
fluorine atom away from triene system has an electrostatic origin (CF/p
repulsion) and is suggested to be a reason for the increase of the reaction
barrier in 19,19-difluorosubstituted model 2.
Acknowledgment. This work was supported by the Osterreichische Nationalbank (Jubiläumsfondsprojekt Nr.: 7395 to W. R.) and supported by the National Science Foundation (CHE-9901661 to R.D.B). We are also thankful to the University of Vienna Computer Center and the National Center for Supercomputing Applications Urbana, Illinois and Lexington, Kentucky for generous amounts of computer time.
References.
1. (a) E. Havinga, Experientia29 (1973) 1181; (b) Akhtar, M.; Gibbons, C. J. J. Chem. Soc.1965, 1101. (c) Moriarty, R. M.; Paaren, H. E. Tetrahedron Lett.1980, 2389. (d) Sialom, B.; Mazur, Y. J. Org. Chem. 1980, 45, 2201. (e) Moriarty, R. M.; Paaren, H. E. J. Am. Chem. Soc. 1981, 46, 970. (f) An exceptionally high thermal unstability was observed for the C19-methoxy and -acetoxy substituted previtamins D. In C19-difluoro previtamin D, no thermal isomerization to the respective vitamin D derivative could be detected.1c The rate constants for thermal [1,7]-H shift are not available.
2. (a) Rodewald, W. J.; Sicinski, R. R. Pol. J. Chem. 1983, 57, 1267. (b) Sicinski, R. R. Acta Chim. Hung. 1992, 129, 191.
3. Breslow, R.; Hoffman, J. H.; Perchonock, C. Tetrahedron Lett.1973, 3723.
4. (a) Saettel, N. J.; Wiest, O. J. Org. Chem. 2000, 65, 2331. (b)Hess, B. A. Jr. J. Org. Chem. 2001, 66, 5897; for the earlier SCF calculations on the [1,7]-antarafacial hydrogen shift in 1,3,5-heptatriene, see Hess, B.A.; Schaad, L.J.;Pancir, J. J.Am.Chem.Soc. 1985, 107, 149. (c) Okamura, W.H.; Elnagar, H.Y.; Ruther, M.; Dobreff, S. J.Org.Chem. 1993, 58, 600.
5. (a) Becke, A. D. Phys. Rev. A 1988, 37, 785. (b) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. 1988, B41, 785. (c) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (d) Stevens, P. J.; Devlin, F. J.; Chablowski, C. F.; Frisch, M. J. J. Phys. Chem.1994, 80, 11623.
6. Michl, J.; West, R. Acc. Chem. Res. 2000, 33, 821.
7. Wienhold, F. in The Encyclopedia of Computational Chemistry. Ed. Schleyer, P.v.R. 1792-1811, John Wiley&Sons, Chichester, 1998.
8. Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986.
9. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98, Revision A.7, Gaussian, Inc., Pittsburgh PA, 1998.
10. (a) Schlegel, H. B. J. Comput. Chem. 1982, 3, 214. (b) Schlegel, H. B. Adv. Chem. Phys. 1987, 67, 249. (c) Schlegel, H. B. In: Modern Electronic Structure Theory, Yarkony, D. R. (Ed), World Scientific: Singapore, 1995, p. 459.
11. (a) Curtiss, L. A.; Raghavachari, K.; Trucks, G. W.; Pople, J. A. J. Chem. Phys.1991, 94, 7221. (b) Curtiss, L. A.; Raghavachari, K.; Pople, J. A. J. Chem. Phys.1997, 106, 1063.
12. Previtamin D has at least eight low-energy (within 2 kcal/mol) ground state conformations that differ in their configuration around single bonds C(5)-C(6) and C(7)-C(8), the sign of their corresponding dihedral angles, and in their A-ring conformations (axial vs. equatorial orientation of 3b -OH): (a) Dauben, W. G.; Funhoff, D. J. H. J. Org. Chem. 1988, 53, 5070. (b) Dmitrenko, O.; Reischl, W. Monatshefte f. Chem.1996, 127, 445. (c) Dmitrenko, O.; Reischl, W. J. Mol Struc. (Theochem)1998, 431, 229. (d) Dmitrenko, O.; Reischl, W.; Vivian, J. T.; Frederick, J. H. J. Mol. Struc. (Theochem) 1999, 467, 195. (c) Dmitrenko, O.; Frederick, J. H.; Reischl, W. J. Mol. Struc.(Theochem)2000, 530, 85.
13. Each system has four transition structures that differ in the A-ring conformation (3b -hydrogen can be axial or equatorial) and helicity of the triene system (right-handed, Pchirality and left-handed, M chirality). To reduce the overall effort and save computational time, we have carried out optimization of models that have the 3b-hydrogen only in the equatorial position. We believe that such a restriction of the A-ring conformational flexibility does not affect significantly the triene portion of the system and relative energetics of the transition structures for the [1,7]-H shift. We optimized 6 transition structures (in 1, 2 and 3 models) and found that TSs with P-helix are always lower in energy by 2.2 kcal/mol in 1 and 2 and 3.6 kcal/mol in 3 at the B3LYP/6-31G(d).
14. Based upon 3-desoxy (3b-H-equatorial)-vitamin D model we estimated reaction exothermicity of DE=4 kcal/mol at the B3LYP/6-31G(d) level of theory. See also ref. 12 for more conformational details of previtamin D and vitamin D molecules.
15. Calculated G2 bond energies are typically 2 kcal/mol higher than experimental BDEs (bond dissociation energies). Bach, R. D.; Dmitrenko, O. J. Am. Chem. Soc., submitted.
16. We failed to locate
GS-2 anion structure in its (+)cZ(+)c conformation to calculate
PA(Ht) as it was done for parent and methoxy- models (GS-1
and
GS-3) since due to electrostatic repulsion in 2, (+)cZ(+)c
geometry was destabilized.