By Robert D. Bach, Colin Thorpe and Olga Dmitrenko
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716.
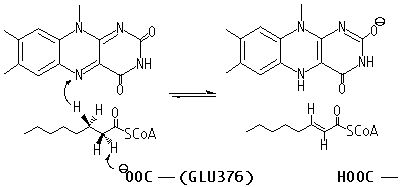
This theoretical study presents new insights into the mechanism of substrate activation in the acyl-CoA dehydrogenases. Removal of the pro-R-a -proton by an active site carboxylate (GLU376 in the medium-chain acyl-CoA dehydrogenase) is accompanied by the concerted elimination of a hydride equivalent to N-5 of the FAD isoalloxazine ring:
We initially address the mechanism of a-proton abstraction without concern for the transfer of a hydride equivalent to the flavin. Computation of the full dehydrogenation reaction, involving the entire isoalloxazine and a minimum suite of ancillary atoms would require extensive computing resources. Further, there is unequivocal experimental evidence for uncoupling a-proton abstraction from reduction of the flavin ring using 3-substituted redox-inactive thioester analogues.1b,1d For example, 3-thiaoctanoyl-CoA has a free pK of about 16 and yet is deprotonated essentially completely when bound to the active site of the acyl-CoA dehydrogenase at pH 7.1b,3
In addition to elevation of the pK of GLU376 by desolvation,1b,1c a critical aspect of this stabilization involves two hydrogen bonds directed towards the thioester carbonyl group.1b,1d A peptide chain N-H group provides one H-bond, and is thus difficult to manipulate experimentally. A second is from the 2'-OH of the ribityl moiety of FAD. Replacement with the deoxy-FAD analog slows the reductive half-reaction (Scheme 1) by about 106-fold and prevents the enolization of 3-thiaoctanoyl-CoA.1d
Our computational approaches are consistent with the critical role played by H-bonds to the substrate carbonyl group.4 However, they provide an additional unanticipated insight, with the identification of a strong ionic C-Hcarboxylate-oxygen H-bond along the reaction coordinate. C-Hoxygen H-bonds have been recognized as important, if under-appreciated, stabilizing interactions in protein crystallography5 and as a directing influence in organic reaction mechanisms. Their role in the deprotonation of carbon acids by carboxylate bases in enzymes has not, to our knowledge, been addressed previously.
In our model studies [constructed in accordance with the X-ray information on acyl-CoA dehydrogenase (MCAD) and butyryl-CoA dehydrogenase (BCAD)], acetate ion (-COOy ) represents the desolvated catalytic base of the enzyme. The H-bond donors to the thioester carbonyl group are methanol (corresponding to the 2'-OH of the FAD) and the N-H of methylformamide (representing the peptide N-H group).
Figure 1 depicts five stages along the reaction coordinate for the proton abstraction step, Table 1 contains the most important thermochemical data.


Figure 1.B3LYP/6-31+G(d,p)
energy diagram6a for a
-proton abstraction model thioester (CH3CH2(C=O)SCH3)
by acetate anion. The lower curve (a) represents reaction energetics
along the reaction coordinate for the system with two H-donors (A-H = CH3O-H
and B-H = H-NCH3(C=O)H). Curve b corresponds to
the model system with one H-donor, methylformamide. The upper curve ( c
) is for a-proton
abstraction without any H-bond donors. TS-2a and TS-2b
are
both first-order saddle points; noproduct minimum (or TS) was located for
curve c. The energy of the product cluster was estimated to be 12.6
kcal/mol above minimum 1c (in the product cluster 3c, CH3CH2(C=O)SCH3CH3COOH,
|OH|=1.01 Å bond was fixed during optimization).6b
Table 1.
Thermochemical Data at the B3LYP/6-31+G(d,p) Level of Theory.a
|
reaction |
kcal/mol |
kcal/mol |
e.u. |
kcal/mol |
|
| 1a-> TS-2a |
|
|
|
|
|
| 1b-> TS-2b |
|
|
|
|
|
a Zero point
energies and statistical mechanical contributions to the entropy are obtained
from the geometries and frequencies calculated at the B3LYP/6-31+G(d,p)
level.
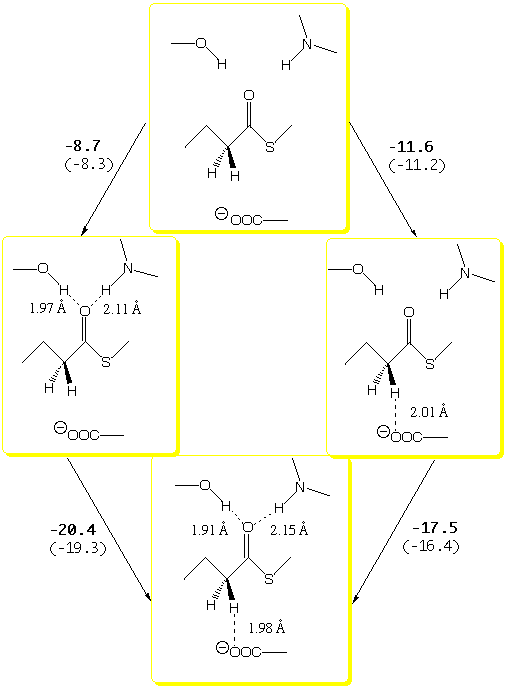
When both H-bonding donors are involved curve a (Fig. 1) is realized. The reactant-cluster 1a, comprising three H-bonding interactions, is 29 kcal/mol lower in energy than its isolated reactants. This large stabilization energy may be conceptually dissected as shown in Scheme 2.7
Scheme 2
If the two H-bonds
to the carbonyl oxygen are formed first, an aggregate stabilization energy
of 8.7 kcal/mol is predicted (typical for neutral species of this type).
However, initial formation of the CHcarboxylate H-bond yields -11.6
kcal/mol. This value is understandably larger than the 1 to 4 kcal/mol
calculated for a number of CHneutral oxygen species8 because
this H-bond is between an electron deficient a-proton
and the carboxylate anion. Obviously, it is immaterial which interaction
forms first upon desolvation of the active site, but note the strong synergy
between them. Thus, the overall stabilization of 29 kcal/mol is 9 kcal/mol
more than the sum of their individual contributions (Scheme 2). This
striking stabilization of the reactant cluster is then reapportioned and
strengthened along the remainder of the reaction coordinate. The
classical activation barrier for a-proton
abstraction by CH3COOy
is only 5.8 kcal (TS-2a) and the product thioenolate
cluster 3a is only 2.4 kcal less stable than reactant
cluster 1a. To our knowledge, TS-2a
is the first example of a first-order saddle point for proton abstraction
adjacent to a carbonyl performed at
a correlated
level.9,10,11 The
a-proton
is more than one-half transferred in the TS. The other changes in geometry
on going from 1ato TS-2a are
minimal with the exception of the H-bond distances (-H ···
O=C) that gradually shorten12 as electron density is transferred
from the carboxylate to the thioester along the reaction coordinate. These
two distances are 1.91 and 2.15 Å in 1a, but are
reduced to 1.74 and 1.84 Å in product cluster 3a
as a consequence of the increase in charge on the thioenolate fragment.13a
 TS-2a
TS-2a
Examination of this change in electron density (ChelpG, MP2/6-31+G(d,p))6a going from 1a to TS-2a is informative. The thioester fragment in 1a has a charge of 0.017 but in TS-2a carries a charge of 0.46. The increase in negative charge on the carbonyl oxygen is only -0.08 (or 17% of the charge transferred to the thioester fragment) going from 1a to TS-2a (consistent with retention of a high degree of carbonyl character in TS-2a; C=O = 1.26Å). Even when the product cluster 3a is examined, the charge on the thioenolate oxygen atom is still only 0.10. Thus negative charge is spread over the entire thioester fragment during enolization and an adequate description of the electrostatic aspects of catalysis should not focus unduly on the carbonyl oxygen.13b
Calculations to mimic removal of the 2'-OH-ribityl H-bond1d (curve b; see earlier) show that the barrier (TS-2b) is raised to 10.4 kcal/mol (Table 1, corresponding to a 103-fold slowing of enolization. In comparison, enolization of the substrate in the absence of both carbonyl H-bonds (curve c) is highly endothermic (with an estimated barrier of 13 kcal/mol).6b Here, multiple approaches failed to locate a bona-fide transition state (first-order saddle point).6b,9,10 The observed tendency of the barrier to increase with an increase of the reaction endothermicity is an evident example of the implementation of the Hammond postulate which works in cases where the ionization potential correlates with bond strengths. A reaction that is highly endothermic (exothermic) is expected to have a high (low) activation barrier and late (early) transition state. A simplified view, consistent with the Hammond postulate that explains the energy diagrams shown in Figure 1 and provides an explanation for the lack of the TS and product in the model c, is given in Figure 2.

Figure 2. Schematic representation of the activation barrier increase and shift in the position of the transition state (to the product side) in models a (two H-donors), b (one H-donor) and c (no H-donors)for a-proton abstraction by acetate anion from model thioester I (CH3CH2(C=O)SCH3). For each model, two curves describe the potential energy of hydrogen atom vs intranuclear distances H-C and H-O. The reduction of the reaction endothermicity (DEreac.) leads to an earlier TS and lower barrier (Hammond postulate). In the case of model c (no H-donors), there is no minimum on the product side for this highly endothermic process and, hence, no TS could be located.
Our results suggest a critical role of H-bonding synergism in the proton abstraction step. The H-bonds to the thioester carbonyl oxygen atom first act to reduce the reaction endothermicity from 16.3 to 2.4 kcal/mol (Figure 2, curve a) due to their larger energetic contribution to the product cluster. In accord with the Hammond postulate and the increased stabilization of the TS versus the pre-reaction complex, a lowering of the reaction barrier and the crossing point of the two surfaces results (Figure 2). Ideally, when the two potential energy wells are equal in energy, the pKs are matched and one has the type of identity-proton transfer. The object of this exercise is to match these pKs as closely as possible in a system that still exhibits a low activation barrier for a-proton abstraction. The stronger the H-donors, the less endothermic the reaction (the better pKa matching), the smaller the barrier and, hence, the earlier the TS. In the absence of the carbonyl H-donors, a reaction with a weakly basic carboxylate will be highly endothermic, the product cluster will be thermodynamically unstable and hence the TS cannot exist.
Figure 1 (curve a) shows that the network of H-bonding interactions stabilize the isolated enolate product by 43 kcal/mol compared to 29 kcal/mol for the thioester reactant. The formidable ability of the dehydrogenase to stabilize enolate-like species by preferential binding has been noted previously.1b-d Importantly, this deep well is not a catalytic trap for normal substrates because they escape via the concerted transfer of a hydride equivalent shown in Scheme 1. Furthermore, migration of the negative charge to the flavin will sharply weaken these hydrogen bonds between enzyme and thioester allowing efficient product dissociation.1b Inspection of Figure 1 and Scheme 2 suggests that placement of a desolvated anionic base in the reaction cluster and the subsequent reapportionment of negative charge and ionic H-bonding interactions over all cluster species13c is a key feature of this enolization reaction.
The presence of water
molecules and their role at the active site of the enzyme catalyzed a-proton
abstraction is a matter of considerable discussion.14 Our model
studies are more consistent with the suggestion that displacement of water
molecules must precede the key proton abstraction step.15 Since
GLU376 is a relatively weak base it seems logical that it would be desolvated
before the deprotonation step involving a weak carbon acid. The basicity
of the carboxylate is greatly increased as it approaches a "naked anion"
upon desolvation and this is likely the process by which the pK of GLU
is increased by some 7 pK units. For example, the proton affinity (PA)
of acetate anion is reduced by 11.2 kcal/mol by the inclusion of just one
H-bonded water molecule (CH3COO· H2Oy).
In Table 2 we summarize the solvent effect estimates (COSMO//B3LYP/6-31+G(d,p))
on the reaction energetics in two model systems a, b.
Table 2.Solvent
Effects on the Reaction Energetics in Model Systems a and b
Calculated at the COSMO//B3LYP/6-31+G(d,p) Level of Theory. Relative Energies
( Erel.) are in kcal/mol.
|
|
gas phase |
e =2.02 |
e =6.89 |
e =78.39 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
These data clearly indicate that an increase in polarity will increasethe reaction barrier. Thus, in a more polar environment, the ground state pre-reaction complex could experience greater stabilization than the corresponding TS or product complex. However, we are aware of the potential effects of local charged residues not directly involved in the reaction, as well as water molecules, and we are going to extend our studies to more complex and comprehensive models.
In sum, these studies
support the importance of electrostatic interactions in the stabilization
of enolate species within the active center of the dehydrogenase. The C-Hcarboxylate
interaction identified here would not only orient the general base prior
to proton abstraction, but also strengthen those H-bonds to the carbonyl
group that prove so critical in the acidification of the a-proton16
and in lowering the transition state for enolization. The synergy between
H-bonding interactions identified here deserves more attention in related
enzymatic examples.
Computational Methods.
Ab initio molecular
orbital calculations were performed with the Gaussian 98 program.6
The Becke three-parameter hybrid functional combined with the Lee, Yang
and Parr (LYP)7 correlation functional, denoted B3LYP was employed in the
calculations using density functional theory (DFT). In all calculations
we used 6-31G(d), 6-31+G(d,p) or 6-311+G(3df,2p) basis sets. The
stationary points on the potential energy surfaces were characterized by
calculations of vibrational frequencies at the B3LYP/6-31+G(d,p) level.
The partial charges were calculated using the NBO and CHelpG methods implemented
in the Gaussian 98 program. Corrections for solvation (single-point COSMO//B3LYP/6-31+G(d,p)energy
calculations) were made using polarizable conductor COSMO model calculations.17
For a design of the
appropriate models for the GLU376 base-catalyzed a-proton
abstraction from CoA, the X-ray structure of the medium chain acyl-CoA
dehydrogenase (MCAD) active site was used as a geometry reference.
Acknowledgments. Supported by the NSF (CHE-9901661) and NIH (GM26643). We thank the National Centers for Supercomputing Applications (Urbana) and the University of Kentucky (Lexington) for generous amounts of computer time.
References
(1) (a) Schopfer, L. M.; Massey, V.; Ghisla, S.; Thorpe, C. Biochemistry 1988, 27, 6599. (b) Thorpe, C.; Kim, J-J. P. FASEB J. 1995, 9, 719. (c) Rudik, I.; Ghisla, S.; Thorpe, C. Biochemistry 1998, 37, 8437. (d) Engst, S.; Vock, P.; Wang, M.; Kim, J-J. P.; Ghisla, S. Biochemistry 1999, 38, 257; and references therein.
(2) Lai, M. T.; Li, D.; Liu, H. W. J. Am. Chem. Soc. 1993, 115, 1619.
(3) Tamaoki, H.; Nishina, Y.; Shiga, K.; Miura, R., J. Biochem. 1999, 125, 285.
(4) (a) Shan, S. O. ; Herschlag, D. Proc. Natl. Acad. Sci. 1996, 93, 14474. (b) Gerlt, J. A.; Kreevoy, M. M.; Cleland, W. W.; Frey, P. A. Chem. Biol. 1997, 4 259. (c) Cleland, W. W.; Frey, P. A.; Gerlt, J. A. J. Biol. Chem. 1998, 273, 25529.
(5) (a) Derewenda, Z.; Lee, L.; Derewenda, U. J. Mol. Biol. 1995, 252, 248. (b) Wahl, M. C.; Sundaralingam, M. TIBS 1997, 22, 97. (c) Bella, J.; Berman, H. M. J. Mol. Biol. 1996, 264, 734; and references therein.
(6) (a) All ab initio calculations used the Gaussian 98 program and all structures were optimized at the B3LYP/6-31+G(d,p) level. Frisch, M. J. et.al.; Gaussian 98, Gaussian, Inc., Pittsburgh PA, 1998. (b) The product cluster does not exist as a minimum at the B3LYP level (and consequently a TS cannot exist by definition) without H-bonding stabilization. Its energy was estimated based upon an optimized cluster with fixed O-H=1.01 Å distance.
(7) The numbers shown in bold are at the B3LYP/6-31+G(d,p) level, whereas numbers in parenthesis are refined by single point B3LYP/6-311+G(3df,2p)//6-31+G(d,p) calculations.
(8) (a) Gu, Y.; Kar, T.; Scheiner, S. J. Amer. Chem. Soc. 1999, 121, 9411. (b) Vargas, R.; Garza, J.; Dixon, D. A.; Hay, B. P. J. Amer. Chem. Soc. 2000, 122, 4750; and references therein.
(9) The unstabilized reaction "isolated reactants-->isolated products" is highly endothermic (16.3 kcal/mol [17.0 at the G2 level of theory]). Highly endothermic a-proton abstraction, if it occurs, would come late along the reaction coordinate and the TS would closely resemble the product. When the base is too weak thermodynamically to effect enolization (without added stabilization) a product cluster can not exist as a minimum. Since a product cluster 3c minimum does not exist at this level6b a product-like structure was used to estimate the barrier. Similarly, highly exothermic a-proton removal typically comes early and can occur without a discernible barrier (TS). Earlier attempts to locate transition structures for a-proton abstraction in highly exothermic systems were successful at the Hartree-Fock level but when electron correlation and ZPE corrections were included, "the transition states disappeared". (a) Perakyla, M. J. Phys. Chem. 1996, 100, 3441. (b) Perakyla, M. J. Chem.Soc., Perkin Trans. 2, 1997, 2185. (c) Perakyla, M. Phys. Chem. Phys. 1999, 1(24), 5643.
(10) For a discussion of problems associated with TSs found at the Hartree-Fock level when the product cluster is not stabilized see: (a) Mulholland, A. J.; Richards, W. G. J. Phys. Chem. 1998, 102(34), 6635.
(11) A TS on the HF/6-31G(d) QM/MM surface for a model citrate synthase reaction corresponding to curve a involving two H-bonds has been reported but no barrier was given. Mulholland, A. J.; Lyne, P. D.; Karplus, M. J. Am. Chem. Soc. 2000, 122(3), 534.
(12) H-bond distances between heteroatoms: 3.14 & 2.89 Å in 1a; 2.94 & 2.79 Å in TS-2a; 2.86 & 2.72 in 3a for N O and O O, respectively.
(13) (a) The charges on the CH3COOy fragment in 1a is 0.88 while the charge on the CH3COOH fragment in 3a is reduced too 0.15. (b) The distribution of developing charge in TS-2a on the CH3CH, carbonyl oxygen and the SCH3 fragments is 64, 17 and 19%. (c) The aggregate charges on the donor fragments in 1a and TS-2a are 0.14 and 0.18.
(14) (a) Sargent, A.L.; Rollog, M.E., Almlof, J.E.; Gassman, P.G.; Gerltt, J.A. J. Mol. Struc. (Theochem) 1996, 388, 145. (b) Warshel, A.; Strajbl, M.; Villa, J.; Florian, J. Biochemistry 2000, 39(48), 14728. (c) Strajbl, M.; Florián, J.; Warshel, A. J. Am. Chem. Soc. 2000, 122, 5354.
(15) (a) Gerlt, J.A.; Gassman, P.G. J.Am.Chem.Soc. 1993, 115, 11552. (b) Gerlt, J.A.; Gassman, P.G. Biochemistry 1993, 32, 11943. (c) Kozarich, J.W.; Gerlt, J.A.; Kenyon, G.L.; Gassman, P.G. J.Am.Chem.Soc. 1991, 113, 9667 (d) Warshel, A.; Papazyan, A.; Kollman P. A. Science 1995, 269, 102.
(16) We believe that presence of flavin in the native enzyme is another additional factor that may significantly contribute to the acidification of the a-proton of acyl-CoA thioesters (Rudik, I.; Thorpe, C. Archives of Biochem. And Biophys. 2001, 392(2), 341). These studies on a larger model (which includes hydride transfer step and flavin molecule) are in progress.
(17) Barone, V.; Cossi,
M.; Tomasi, J. J. Comp. Chem. 1998,19, 404.