Effect of a Charge-Transfer Interaction on the Catalytic Activity of Acyl-CoA-Dehydrogenase.
by Robert D. Bach, Colin Thorpe and Olga Dmitrenko*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716.
Keywords: Acyl-CoA-Dehydrogenase, charge-transfer complex, TD-DFT, proton abstraction,
In order to examine the role of charge transfer (CT) interactions in the catalytic activity of acyl-CoA dehydrogenase, model systems for the reactant complex of tricyclic or bicyclic models of FAD (Flox) with the thioenolate portion of acyl-CoA were studied. Singlet excitation energies have been estimated at the TD-B3LYP/6-31G(d)//B3LYP/6-31+G(d,p) level of theory. Location and intensity of the charge-transfer band in the UV-absorption spectrum of the complex depends on the distance between model substrate and the isoalloxazine ring and becomes prominent at approximately van der Waals contact. The pre-reaction complex for hydride transfer to flavin [fully optimized at B3LYP/6-31+G(d,p)] possesses a sandwich-like structure with a contact distance of about 3 Å between participants (which is close to van der Waals contact) and has about 0.7 electrons transferred to flavin. We suggest that the formation of CT complex in the enzyme has a significant impact on the barrier to a -proton abstraction: it reduces the a -proton affinity of the thioester, thereby lowering the activation energy for the first step in this enzymatic process.
1. Introduction
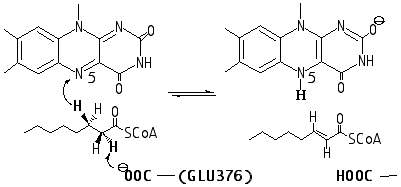
Acyl-CoA dehydrogenases catalyze the two-electron oxidation of a broad range of fatty acids with the conversion of acyl-CoA thioesters to their corresponding a , b -enoyl-CoA products. Dehydrogenation occurs by the breakage of two kinetically stable C–H bonds of the substrate introducing a carbon-carbon double bond as shown in Scheme 1.1 The first C–H bond breaking requires chemical activation of an adjacent thioester functionality and abstraction of hydrogen as H+ by the weakly basic Glu376-COOy with, what is presumed to be, a concomitant expulsion of a b -hydride that is transferred to N-5 of the FAD isoalloxazine ring. Strong support for this concerted pathway, as opposed to the intervention of a discrete intermediate such as a delocalized a -carbanion, comes from kinetic isotope effect (KIE) studies.1
Scheme 1
Our recent computational study2 has suggested that placement of a desolvated anionic base in the reaction cluster and the subsequent reapportionment of negative charge and ionic H-bonding interactions over all cluster species is a key feature of this enolization reaction. It has been found that H-bonds to the carbonyl oxygen of the enolate significantly reduce the proton affinity of the a -proton (thereby providing matching of pKa) and decrease in the reaction endothermicity that, in line with the Hammond postulate, results in the lowering of the reaction barrier for the a -proton abstraction.
Analysis of the X-ray structure of the medium-chain acyl-CoA3 dehydrogenase (MCAD) shown in Figure 1 has revealed the potential importance of sandwich-like arrangement of the substrate and
isoalloxazine ring system of FAD. A number of experimental studies have provided spectroscopic evidence for the formation of a charge-transfer complexes between FAD and CoA analogs.4 Recently, Rudik and Thorpe raised the possibility that charge-transfer (CT) interaction between the developing enolate and the flavin chromophore contribute to the stabilization of the transition state during the dehydrogenation of CoA-thioester substrates.5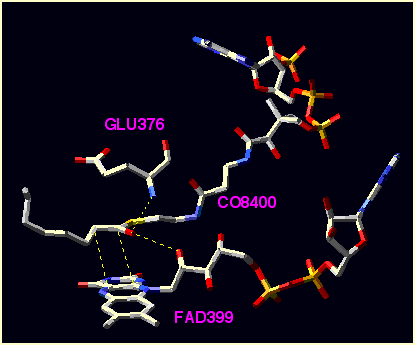
Figure 1. Substrate and selected residues of the X-ray structure for medium-chain acyl-CoA3 dehydrogenase .
In this paper we address the mechanism of a -proton abstraction in the presence of a CT interaction without concern for the concomitant transfer of a hydride equivalent to the flavin. There is unequivocal experimental evidence for uncoupling a -proton abstraction from reduction of the flavin ring using 3-substituted redox-inactive thioester analogues.1b,1d It is not our intent to represent the enzyme itself, but rather to provide a model system that serves to describe the intrinsic gas phase behavior to illustrate how flavin proximity affects the activation barrier for a -carbonyl proton removal.
2. Computational details
Quantum chemistry calculations were carried out using the Gaussian98 program system6 utilizing gradient geometry optimization.7 All geometries were fully optimized using the B3LYP functional8,9 with 6-31G(d) and 6-31+G(d,p) basis sets. Vibrational frequency calculations at the B3LYP/6-31G(d) level of theory were performed to characterize the stationary points as either minima or transition structures (first-order saddle point). Proton affinities (PA) were estimated as the difference in total energy between the protonated and the corresponding neutral species at the B3LYP/ 6-31+G(d,p) level of theory. It has been shown that such an estimation produces PAs that are systematically lower by 3-5 kcal/mol than experimental PAs values.2 Unless otherwise stated all energy values quoted in the text are at the B3LYP/6-31+G(d,p) level. Vertical energies of the excited state transitions have been calculated using the TD B3LYP/6-31G(d) time-dependent DFT method.10 Corrections for solvation were made using polarizable conductor COSMO model calculations.11
3. Models.
In our extended models based upon a fragment of the active site of the X-ray structure for medium-chain acyl-CoA dehydrogenase, 3 thioester CH3CH2CH2(C=O)SCH3 was chosen to model the CoA substrate. The active site base (-COOy ) and the amide hydrogen donor (H-N<) are combined in a single molecule (H(C=O)-NH-CH2CH2CH2COOy )) which more closely resembles the GLU376 side chain. To model FAD, we used a tricyclic oxidized flavin molecule (Flox) with the ribityl moiety represented by a —CH2CH2OH group which may hydrogen bond to the substrate carbonyl oxygen. In some qualitative calculations, we used smaller model compounds, bicyclic flavin (pterin derivative) and CH3CH2(C=O)SCH3.
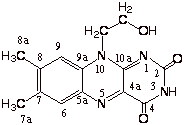 Flox
Flox
4. Results and Discussion
4.1. Effect of flavin proximity on the a -proton affinity of thioester
In our recent study we have demonstrated an important role of the carbonyl H-bonds for a -proton abstraction. Their major contribution to the catalysis is lowering the pKa of the a -proton2. The effect of H-bond donors on the relative stability of the enolate ion resulting from a -proton abstraction was predicted from their respective proton affinities (Table 1) estimated from B3LYP/6-31+G(d,p) total energies. Naked thioenolate anion, [CH3CH(C=O)SCH3]-, has a much higher PA than acetate anion (367.9 vs 352.9 kcal/mol), which means, that in the absence of H-donors, acetate anion is too weak a base to abstract an a -proton. For this reason, neither transition structure nor product cluster have been located2. Two H-donors to the model substrate reduce the a -proton affinity to the value 345.3 kcal/mol which is 7.6 kcal/mol lower than PA of the base acetate anion. The effect of one H-donor, methylformamide, is not so pronounced; the PA of acetate anion is slightly lower (by 1.4 kcal/mol) than the PA of CH3CH(C=O)SCH3• HNCH3(C=O)H. This H-bond mediated reduction of the energy needed for a -proton abstraction from the thioester leads to a significant decrease (from 10.4 to 5.8 kcal/mol) in the reaction barrier (Scheme 2).
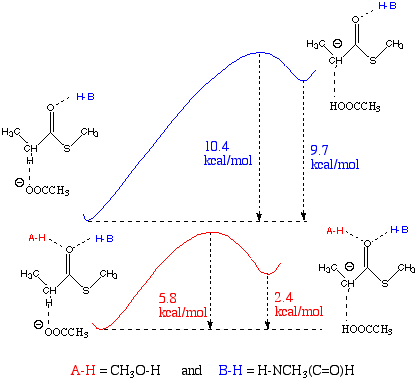
Scheme 2
Table 1. Estimated Proton Affinities (PA) of Anionic Bases and Enolate Intermediates Based upon B3LYP/6-31+G(d,p) Total Energies.
|
anion |
PA (kcal/mol) |
|
|
CH3COO |
} bases |
352.9 |
|
(CHO)NHCH2CH2CH2COOc (all trans) |
345.1 |
|
|
CH3CH(C=O)SCH3 |
369.2 |
|
|
CH3CH2CH(C=O)SCH3 |
367.9 |
|
|
CH3CH(C=O)SCH3• HNCH3(C=O)H |
354.3 |
|
|
CH3CH(C=O)SCH3•(CH3OH, HNCH3(C=O)H) |
345.3 |
|
|
CH3CH2CH(C=O)SCH3• CH3OH |
359.1 |
In order to get an initial idea on how the flavin proximity found in the X-ray active site structure may affect the a -proton abstraction, we calculated proton affinities for a simplified sandwich-like system consisting of thioenolate,CH3CH(C=O)SCH3, and a bicyclic oxidized flavin model (Figure 2). The distance (d, Å) was varied between 3 and 5.0 without post-optimization. The starting complexes (neutral and anionic) were pre-optimized at the B3LYP/6-31+G(d,p) with the two C-C distances (see Figure 2) fixed to 3 Å and two dihedral angles fixed to +/-90° in order to keep the planes of the flavin [defined by {N5,C4a,C10a}] and substrate [defined by {O=C1Ca }] parallel.
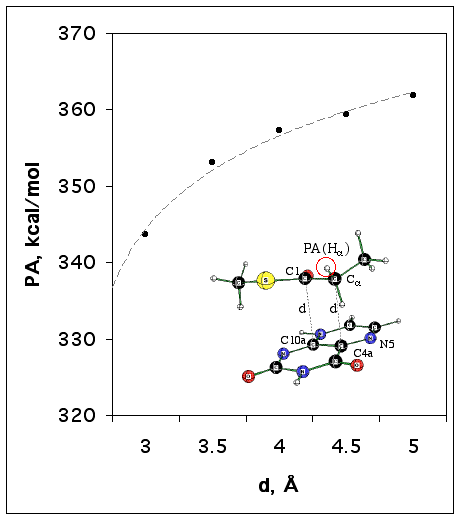
Figure 2. The dependence of a -proton affinity for CH3CH(C=O)SCH3/Flox model anionic complex (shown in insertion) on the distance to flavin (d) estimated at the B3LYP/6-31+G(d,p) level of theory.
The results of these estimations are presented in Figure 3. They suggest that the proximity of the flavin to the substrate may have a strong acidifying effect upon the a -hydrogen of the thioester, so that bases even weaker than acetate anion can be effective.
4.2. The a -proton abstraction in model systems with and without flavin
Using the fragment of the enzyme active site X-ray structure we selected a more extended base (CHO)NHCH2CH2CH2COO to mimic GLU376. Its PA was estimated to be 345.1 kcal/mol (Table 1). Thus, comparing this value with the proton affinities given in Table 2, one may expect a significant reduction of the reaction barrier when a flavin molecule is docked in a sandwich-like structure with the distance d=3 Å (which is slightly less then sum of van der Waals radii of two carbons).
First, we found that the reaction barrier for the a -proton abstraction in the model system (Figure 3) without flavin is 7.7 kcal/mol.
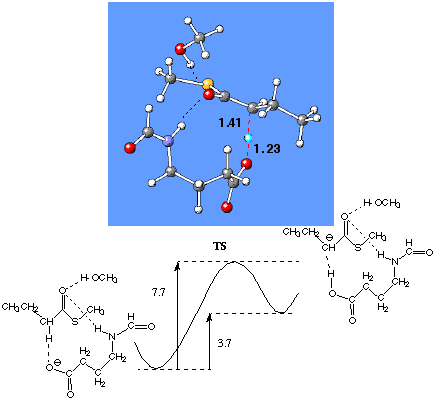
Figure 3. Fully optimized transition structure and relative energies (kcal/mol) of the optimized pre-reaction cluster for thioester, the TS and product cluster of model system {CH3CH2CHH(C=O)SCH3•(CH3OH,(H(C=O)-NH-CH2CH2CH2COOy )} at the B3LYP/6-31+G(d,p) level of theory.
Then the tricyclic oxidized flavin molecule was placed with the orientation appropriate to the crystal structure of the enzyme. The pre-reaction complex, transition structure and product complex have been optimized with two C-C distances (C1-C10a and Ca -C4a) constrained to 3 Å. The resulting reaction barrier was found to be about 2-fold lower (3.6 kcal/mol, Figure 4). The B3LYP/6-31G(d) calculations performed for the system with contact distance d=4Å, result in the reaction barrier 2.3 kcal/mol higher than that for the d=3Å. Remarkably, the 3Å-constrained pre-reaction complex is about 12 kcal/mol higher in energy than the 4Å-constrained pre-reaction complex. Thus, one may definitely conclude that flavin proximity lowers the reaction barrier for a -proton abstraction. The increase of contact distance by 1 Å may result in the 50-fold decrease in the reaction rate.
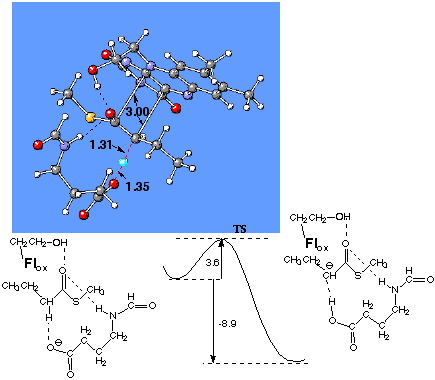
Figure 4. The transition structure optimized at the B3LYP/6-31+G(d,p) level for the a -proton abstraction reaction in model system of CoA-dehydrogenase with docked oxidized flavin. The distance between Flox and substrate is fixed to 3 Å. The reaction barrier and reaction energy (kcal/mol) are at the B3LYP/6-31+G(d,p) level of theory.
4.3. Charge-transfer thioenolate/Flox complexes and their spectral characteristics
The anionic form of thioester substrate and oxidized flavin form a stable sandwich-like complex without any geometrical constraint (Figure 5). The estimated total energy of complexation is 22.2 kcal/mol. When the ribityl moiety, represented by the —CH2CH2OH group, is substituted by CH3 (lumiflavin) in order to prevent the contribution of the H-bond stabilization, the complexation energy drops to 16.3 kcal/mol, which we assign to the charge-transfer interaction energy. In spite of the decrease in complexation energy upon H-bond removal, the interacting flavin and thioenolate come into closer contact [C1C10a=3.111 Å Ca C4a=2.897Å vs. 3.269 Å and 2.934 Å, Figure 5]. Interestingly, when the thioester substrate is replaced by the 3-thia-butanoyl-CoA model substrate (Figure 6), the complexation energy is reduced significantly [22.2 vs 15.6 kcal/mol, Figure 5(A) and Figure 6], and the distance between substrate and flavin is markedly increased.
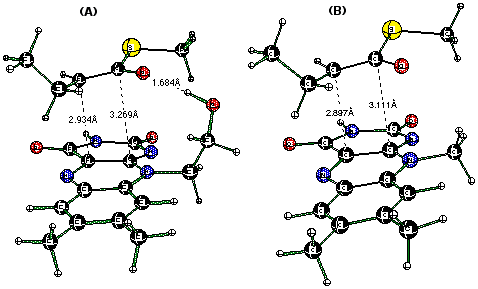
Figure 5. CH3CH2CH(C=O)SCH3-/Flox (anionic form of the substrate/flavin) complexes with the ribityl moiety represented by —CH2CH2OH (A) and CH3 (B) groups optimized at the B3LYP/6-31+G(d,p) level of theory.

Figure 6. CH3SCH(C=O)SCH3-/Flox complex optimized at the B3LYP/6-31+G(d,p) level of theory. The total energy of complexation (including OH…O=C< bonding) is 15.6 kcal/mol.
Spectroscopic studies of the acyl-CoA dehydrogenase model with the redox-inactive thioester analog 3-thia-octanoyl-CoA (that cannot do the hydride transfer to the flavin) have indicated the appearance of an intense charge-transfer band (centred at 800 nm) generated upon removal of the weakly acid a -proton.4,5
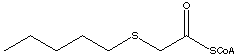
We performed TD DFT (B3LYP) calculations on the charge-transfer model complex with the CH3SCH(C=O)SCH3- model anionic form of the substrate (Figure 6). This charge transfer complex has three major absorption bands characterized by the calculated Franck-Condon (FC) transitions at 957, 408 and 337 nm [TD B3LYP/6-31G(d)//B3LYP/6-31+G(d,p),Table 2]. The first transition can be assigned to a charge-transfer band, whereas the two last transitions belong to flavin as indicated by the summary of TD B3LYP calculations for flavin (Table 3). We also examined the solvent effect (COSMO, Table 2) and found no significant changes except some increase in the oscillator strengths. For a comparison, the absorption spectrum of lumiflavin in water shows two characteristic bands at 446 nm and 370 nm, followed by an intense band at 270 nm.12 Thus, as also noted in ref. 13, the theoretically predicted spectrum is somewhat blue-shifted with respect to the experimental one.
It should be noted that generally the TD DFT (B3LYP) approach predicts the excitation energies for low-lying excited states for single molecules remarkably well (very often within a few nm). As indicated in Table 2, the predicted CT band position is highly dependent upon the level of theory at which the complex has been optimized. The complex optimized with smaller basis set (6-31G(d)) is tighter and has a shorter wavelength of the CT transition (859 nm), which better agrees with the experimental observation. Since we do not include in our model all the residues surrounding the substrate/Flox complex in studies of the acyl-CoA dehydrogenase with the redox-inactive thioester analog 3-thia-octanoyl-CoA,5 one should not necessary expect excellent agreement with experiment. The contact distance in the CT complex can be affected by external steric and electrostatic interactions.
Table 2. Singlet allowed FC transitions (nm) calculated for the CH3SCH2(C=O)SCH3/Flox complex optimized at the B3lYP/6-31G(d) level using the time-dependent DFT method [TD(Nstates=12) B3LYP/6-31G(d)]. Numbers 957, 408 and 337 correspond to complex optimized at B3LYP/6-31+G(d,p) level, in which CT-partners are at longer contact distance. Oscillator Strengths (f) are given in parentheses. Calculations for tetrahydrofuran (THF) as solvent were performed with the COSMO model.
|
State |
Gas-phase |
THF |
|
S1 (HOMO-LUMO) |
957 nm (f=0.11) 859 nm (f=0.13) |
838 nm (f=0.11) |
|
S6 |
408 nm (f=0.11) 402 nm (f=0.12) |
407 nm (f=0.13) |
|
S10 |
337 nm (f=0.05) 326 nm (f=0.05) |
327 nm (f=0.10) |
Table 3. Low-energy singlet allowed FC transitions (nm) calculated for tricyclic Flox optimized at the B3lYP/6-31+G(d,p) level using the time-dependent DFT method [TD(Nstates=12) B3LYP/6-31G(d)]. Oscillator Strengths (f) are given in parentheses. Calculations for tetrahydrofuran (THF) as solvent were performed with the COSMO model.
|
transition |
Gas phase |
THF |
Water |
|
HOMOà LUMO |
410 nm (f=0.10) |
410 nm (f=0.15) |
413 nm (f=0.14) |
|
HOMO-3à LUMO |
341 nm (f=0.13) |
337 nm (f=0.16) |
345 nm (f=0.17) |
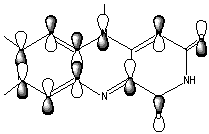
The LUMO orbital of flavin consist of bonding/antibonding interactions of p (C8C7), p (C9C9a), lp(N10), p *(C4aN5), p *(C10aN1), p *(C2=O), p *(C4=O) orbitals (Figure 7).
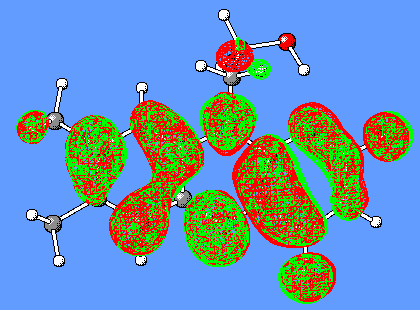
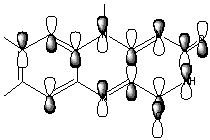
Figure 7. LUMO orbital of Flox [B3LYP/6-31G(d)].
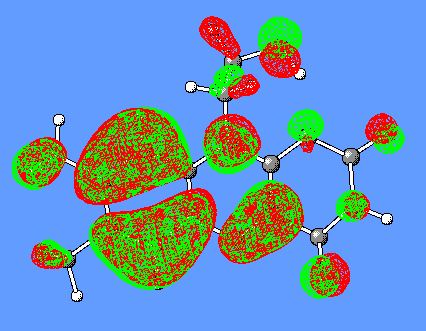
The HOMO and (HOMO-3) molecular orbitals are mostly different combinations of p orbitals (Figure 8).
 HOMO
HOMO
p (C6C7C8),p (C5aC9aC9),p (C4aN10),lp(N1),lp(OC2),lp(OC4)
 HOMO-3
HOMO-3
p (C7C6C5a),p (C8C9C9a),p (C4aN5),lp(N10), lp(OC4), lp(OC2, lp(N1), lp(N3)
Figure 8. HOMO and (HOMO-3) orbitals of Flox [B3LYP/6-31G(d)].
The LUMO orbital of the oxidized flavin (Figure 8) has a clearly distinguished p character of the C4a-C10a bond. We suggest that this may result in a charge-transfer interaction (partial electron transfer) between the substrate and Flox, in particular, if the substrate has a complementary p bond with an excess of electrons.
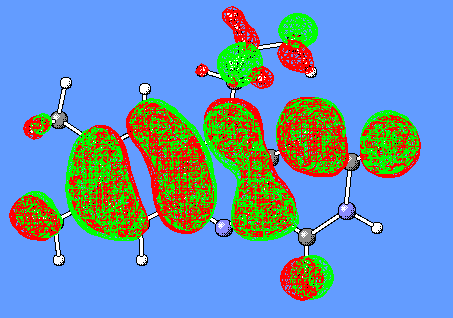
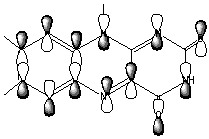
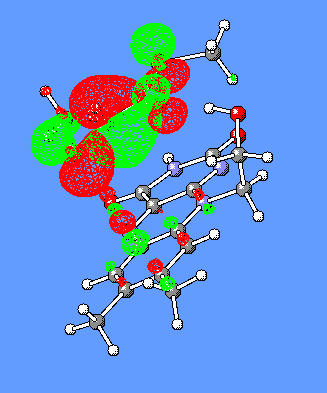
In the CH3SCH(C=O)SCH3-/Flox model complex discussed above (Figure 6), the HOMO and LUMO orbitals responsible for generation of the CT band are the corresponding HOMO and LUMO of isolated CH3SCH(C=O)SCH3- and Flox(Figure 9).
The same observation was made for CH3CH2CH2(C=O)SCH3/Flox (substrate/flavin) complexes which have been optimized with and without constraint of the C1C10a and Ca C4a distances. The CH3CH2CH(C=O)SCH3/Flox complex optimized without constraints is shown in Figure 5(A). Then, distances d= C1C10a= Ca C4a were fixed to 3 Å and re-optimized. Two other complexes with the distances increased to 3.5 and 4 Å were taken for excited state energy calculation without preliminary optimization. The results summarized in Table 4 suggest that the CT band moves to the low-energy region and loses its intensity with increase in contact distance d, whereas the two other flavin bands are not affected so significantly. The decrease in contact distance leads to an increase in negative charge on Flox up to about —0.7 e in the CT complex which is 70% of whole "-1" charge of the complex initially provided by thioenolate.
Table 4. Singlet allowed FC transitions (nm) calculated for the CH3CH2CH(C=O)SCH3/Flox complexes [charge-transfer CT complex has been fully optimized at the B3lYP/6-31+G(d,p) level] using the time-dependent DFT method [TD(Nstates=12) B3LYP/6-31G(d)]. The estimated Mulliken charge (electrons) on Flox is given in the last row.
|
CT |
3Å |
3.5Å |
4Å |
|
644 nm (f=0.23) |
645 nm (f=0.22) |
961 nm (f=0.15) |
1390 nm (f=0.10) |
|
395 nm (f=0.11) |
392 nm (f=0.10) |
410 nm (f=0.13) |
418 nm (f=0.13) |
|
321 nm (f=0.05) |
323 nm (f=0.04) |
326 nm (f=0.08) |
324 nm (f=0.08) |
|
Charge transferred to Flox |
|||
|
-0.66 |
-0.61 |
-0.37 |
-0.33 |
 HOMO
HOMO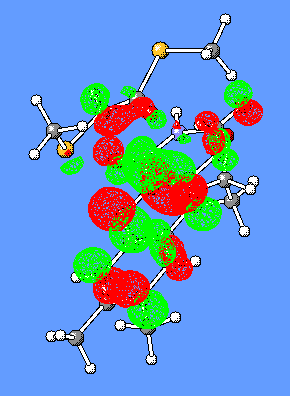 LUMO
LUMO
Figure 9. HOMO and LUMO orbitals for the CH3SCH2(C=O)SCH3/Flox (substrate/flavin) complex responsible for the CT-complex formation and its absorbance at the long-wavelength region.
5. Conclusions.
The present computational study, in conjunction with available published experimental data on acyl-CoA-dehydrogenases, suggests that the developing enolate can be subjected to strong charge-transfer interaction with flavin (16.3 kcal/mol). Analysis of the charge distribution in thioenolate/Flox complexes indicates significant charge transfer to flavin (about 70%). This type of complex is characterized by an intense charge-transfer absorption band (644 nm) which is assigned to p à p * transfer from the highest occupied molecular orbital of thioenolate to the lowest virtual orbital of flavin. The energy of this charge-transfer vertical (Franck-Condon) excitation depends strongly on the contact distance but appears less sensitive to the polarity of microenvironment.
The B3LYP calculations of two model systems (with and without flavin) designed for simulation of a -proton abstraction in acyl-CoA-dehydrogenases show that proximity of flavin may have a strong impact on the reaction efficiency leading to a 2-fold decrease in the reaction barrier. It is suggested that flavin proximity due to a charge-transfer interaction causes a significant acidifying effect on the a -proton.
The available X-ray structures of acyl-CoA-dehydrogenases suggest that the geometrical constraint imposed by the active site of the enzyme as well as H-bonding with the substrate carbonyl oxygen forces the thioester-substrate to approximate within van der Waals contact with the flavin prosthetic group. This, according to our computational simulations, should acidify the a -proton of the substrate and accelerate base-catalyzed proton abstraction.
The effect of flavin proximity on the a -proton abstraction step as well charge-transfer interaction between thioenolate and flavin raises the possibility of a significant impact upon the reaction-dependent molecular motion on the overall reaction kinetics.
Acknowledgment. This work was supported by the National Science Foundation (CHE-0138632), by NIH GM26643 and partially supported by National Computational Science Alliance under CHE990021N and utilized the NCSA SGI Origin2000 and University of Kentucky HP Superdome.
References.
1. (a) Schopfer, L. M.; Massey, V.; Ghisla, S.; Thorpe, C. Biochemistry 1988, 27, 6599. (b) Thorpe, C.; Kim, J-J. P. FASEB J. 1995, 9, 719. (c) Rudik, I.; Ghisla, S.; Thorpe, C. Biochemistry 1998, 37, 8437. (d) Engst, S.; Vock, P.; Wang, M.; Kim, J-J. P.; Ghisla, S. Biochemistry 1999, 38, 257; and references therein. (e) Johnson, J. K.; Srivastava, D. K. Biochemistry 1993, 32, 8004.
2. Bach, R.D.; Thorpe, C.; Dmitrenko, O. J.Phys.Chem.A. 2002, 106, 4325-4335.
3. (a) Kim, J-J.P; Wang, M.; Passchke, R. Crystal structures of medium chain acyl-CoA dehydrogenase from pig liver mitochondria with and without substrate. Proc. Natl. Acad. Sci. USA 1993, 90, 7523. (b) Djordjevic, S.; Pace, C. P., Stankovich, M. T.; Kim, J. J. Biochemistry 1995, 34, 2163; (c) PDB Ref.Code is 3MDE for medium-chain Acyl-CoA dehydrogenase and 1BUC for butyryl-CoA dehydrogenase; Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Research, 2000, 28. 235.
4. (a) Lau, S.-M.; Brantley, R.K.; Thorpe, C. Biochemistry 1988, 27, 5089. (b) Tamaoki, H.; Nishina, Y.; Shiga, K.; Miura, R. J. Biochem. 1999, 125, 285.
5. Rudik, I.; Thorpe, C. Arch. Biochem. Biophys. 2001, 392 (2), 341.
6. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone,V.; Cossi, M.; Cammi,R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Baboul, A. G.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W; Johnson, B.; Chen, W.; Wong, M. W.; Andres, J. L.; Gonzalez, C.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian 98, Revision A.7, Gaussian, Inc., Pittsburgh PA, 1998.
7. (a)
Schlegel, H. B. J. Comput. Chem. 1982, 3, 214-218. (b) Schlegel, H. B. Adv. Chem. Phys. 1987, 67(Pt. 1), 249-286. (c) Schlegel, H. B. In Modern Electronic Structure Theory, Yarkony, D. R., Ed.; World Scientific: Singapore, 1995; pp 459.8. (a)
Becke, A. D. Phys. Rev. A 1988, 38, 3098-3100. (b) Lee, C.; Yang, W. and Parr, R. G. Phys. Rev. B 1988, 37, 785-789.9. (a) Becke, A. D. J. Chem. Phys. 1993, 98, 5648-5652. (b) Stevens, P. J.; Devlin, F. J.; Chabalowski, C. F. and Frisch, M. J. J. Phys. Chem. 1994, 98, 11623-11627.
10. (a) Bauernschmitt, R. ; Ahlrichs R. Chem. Phys. Lett. 1996, 256, 454—464. (b) Casida, M. E.; Jamorski, C.; Casida, K. C.; Salahub, D. R. J. Chem. Phys. 1998,108, 4439. (c) Stratmann, R. E.; Scuseria, G. E. ; Frisch, M. J. J. Chem. Phys. 1998, 109, 8218—8224.
11. Barone, V.; Cossi, M.; Tomasi, J. J. Comp. Chem. 1998,19, 404.
12. Dudley, K. H.; Ehrenberg, A. ; Hemmerich P. ; Müller F. Helv.
Chim. Acta 1964,47,1354—1383.
13. Neiß, C.; Saalfrank, P. ; Parac M.; Grimme S. J Phys. Chem A. 2003,107: (1) ,140-147.